Reaktionsmechanismen
Aldoladdition
*Arndt-Eistert-Synthese
*Baeyer-Villiger-Reaktion
*Balz-Schiemann-Reaktion
*Barton-Reaktion
*Beckmann-Umlagerung
*Benzoin-Addition
*Birch-Reduktion
*Bischler-Napieralski-Reaktion
*Biuret-Reaktion
*Blanc-Reaktion
*Bohn-Schmitt-Reaktion
*Bouveault-Blanc-Reduktion
*v. Braun-Reaktion
*Bucherer-Reaktion
*Cannizzaro-Reaktion
*Claisen-Kondensation
*Claisen-Umlagerung
*Clemmensen-Reduktion
*Cope-Eliminierung
*Cope-Umlagerung
*Corey-House-Synthese
*Corey-Seebach-Methode
*Criegee-Oxidation
*Curtius-Abbau
*Demjanow-Reaktion
*Diaza-Cope-Umlagerung
*Dieckmann-Kondensation
*Diels-Alder-Reaktion
*Doebner-v.Miller-Chinaldinsynthese
*Edman-Abbau
*Einhorn-Verfahren
*Eschweiler-Clarke-Methylierung
*Finkelstein-Reaktion
*Fischersche-Indolsynthese
*Fischer-Tropsch-Synthese
*Frank-Caro-Verfahren
*Friedel-Crafts-Acylierung
*Vollhardt S.647 und S.715
*
Friedel-Crafts-Alkylierung
*Friedländer-Synthese
*Fries-Umlagerung
*Gabriel-Synthese
*Gattermann-Aldehydsynthese
*Gattermann-Koch-Synthese
*Gomberg-Bachmann-Reaktion
*Graebe-Ullmann-Synthese
*Grignard-Reaktion
*Hantzsche-Dihydropyridinsynthese
*Hantzsche-Thiazolsynthese
*Haworth-Synthese
*Hell-Volhard-Zelinsky-Reaktion
*Heumannsche-Indigosynthese
*Hinsberg-Reaktion
*Hoesch-Ketonsynthese
*Hofmann-Abbau
*Hofmann-Alkylierung
*Hofmann-Eliminierung
*Horner-Emmons-Reaktion
*Houndry-Verfahren
*Huang-Minlon-Methode
*Hunsdiecker-Reaktion
*Iwanow-Reaktion
*Knoevenagel-Reaktion
*Knorrsche-Pyrrolsynthese
*Koch-Haaf-Reaktion
*Kolbe-Nitrilsynthese
*Kolbe-Schmitt-Synthese
*Kolbe-Synthese
*Kupplungsreaktionen
*C-Kupplung
*N-Kupplungen
*
Leuckart-Wallach-Reaktion
*Lobry de Bruyn-vanEkenstein-Umlagerung
*Lossen-Abbau
*Madelung-Synthese
*Mannich-Reaktion
*McLafferty-Umlagerung
*McMurry-Reaktion
*Meerwein-Ponndorf-Verley-Reduktion
*Meerwein-Schuster-Reaktion
*Michael-Addition
*Michaelis-Arbusow-Reaktion
*Müller-Rochow-Synthese
*Nef-Reaktion
*Nesmejanow-Reaktion
*Oppenauer-Oxidation
*Paal-Knorr-Synthese
*Paterno-Büchi-Reaktion
*Perkin-Synthese
*Peterson-Olefinierung
*Prileschajew-Reaktion
*Prins-Reaktion
*Pschorr-Synthese
*Pummerer-Umlagerung
*Raschig-Verfahren
*Reformatsky-Reaktion
*Reimer-Tiemann-Synthese
*Reppe-Synthesen
*Reppe-Carbonylierung
*Reppe-Cyclisierung
*Reppe-Ethinylierung
*Reppe-Vinylierung
*
Sachsse-Bartholomé-Verfahren
*Sandmeyer-Reaktion
*Schiemann-Reaktion
*Schmidt-Reaktion
*Schotten-Baumann-Verfahren
*Shapiro-Reaktion
*Simmons-Smith-Reaktion
*Skraupsche-Synthese
*Sommelet-Reaktion
*Stephen-Reduktion
*Stobbe-Kondensation
*Strecker-Synthese
*Swarts-Reaktion
*Swern-Oxidation
*Tischtschenko-Reaktion
*Traube-Synthese
*Tschitschibabin-Reaktion
*Twitchell-Verfahren
*Ullmann-Reaktion
*Vilsmeier-Haack-Synthese
*Wacker-Hoechst-Verfahren
*Wacker-Verfahren
*Wagner-Meerwein-Umlagerung
*Willgerodt-Kindler-Reaktion
*Williamson-Synthese
*Wittig-Reaktion
*Wöhlersche-Harnstoffsynthese
*Wohl-Verfahren
*Wohl-Ziegler-Reaktion
*Wolff-Kishner-Reduktion
*Wolff-Umlagerung
*Wurtz-Fittig-Synthese
*Wurtz-Synthese
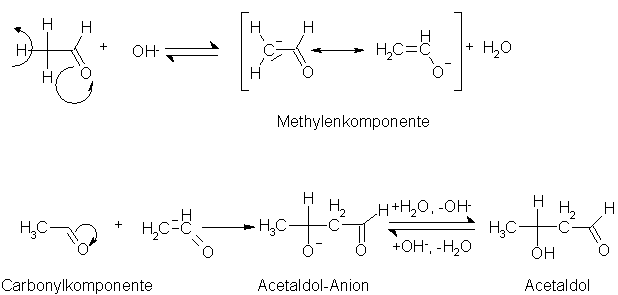
*Früher wurde die Aldoladdition auch als Aldolkondensation bezeichnet, was nicht richtig ist, da kein Wasser abgespalten wird. Die Bezeichnung Aldol ist eine Abkürzung für ALDehydalkohOL, sie zeigt auch die Doppelfunktion solcher Verbindungen an. Aldehyde, die eine zur Carbonylgruppe
a-ständige Methyl- oder Methylengruppe tragen, dimerisieren unter Säuren- oder Basenkatalyse (verdünnte Alkalihydroxide, Alkalicarbonate, Alkalicyanide, Natriumacetat, verdünnte Salzsäure) zu b-Hydroxyaldehyden (Aldolen). Der Mechanismus der basenkatalysierten Aldoladdition, wird wie folgt beschrieben. Das Hydroxidion spaltet das durch den -I-Effekt der CO-Gruppe aktivierte Wasserstoffatom der Methylgruppe ab. Es entsteht so ein, durch Mesomerie stabilisiertes, Carbanion, welches mit dem einsamen Elektronenpaar an das positivierte C-Atom eines anderen Carbonylmoleküls addiert. Dadurch entsteht als Vorstufe ein Aldolanion, welches als starke Base durch das Wasser zum Aldol protoniert wird. Da die gesamte Aldoladdition eine reine Gleichgewichtsreaktion ist, hängen die Ausbeuten der Produkte stark von den Reaktionsbedingungen (z.B. pH-Wert) ab.
Beyer/Walter S.200
Organikum S.467
Vollhardt S.774
Vorlesung
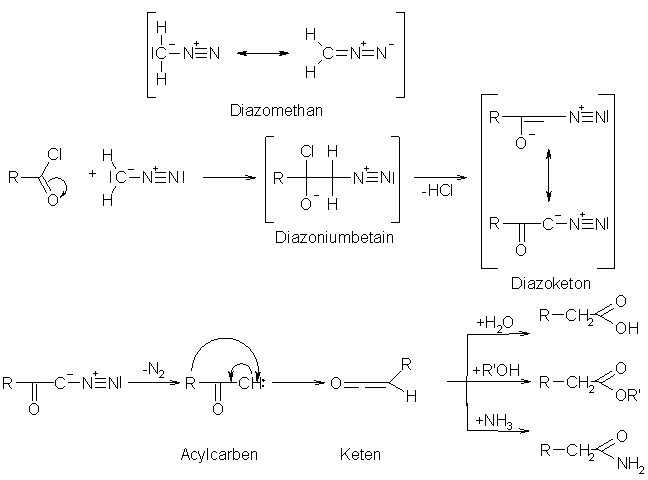
Umsetzung eines Carbonsäurechlorids mit Diazomethan. Dabei kommt es zu einer Acylierung am C-Atom. Intermediär entsteht ein Diazoniumbetain, das sofort Chlorwasserstoff abspaltet und ein mesomeres Diazoketon bildet. Die Reaktion folgt einem Additions-Eleminierungs-Mechanismus. Das Diazoketon spaltet bei höheren Temperaturen, in Gegenwart von Metallkatalysatoren, Stickstoff ab. Dabei wird eine Elektronenmangelverbindung gebildet, ein Carben. Durch eine Wolff-Umlagerung stabilisiert sich das Carben unter Ausbildung einer Doppelbindung zu einem Keten.
Setzt man das Keten mit :
Wasser um, wird eine Carbonsäure gebildet.
einem Alkohol um, wird ein Carbonsäureester gebildet.
Ammoniak um, wird ein Carbonsäureamid gebildet.
 Beyer/Walter
S.254
Beyer/Walter
S.254
Organikum S.603
Vorlesung
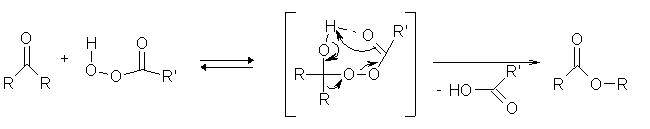
Bei dieser Reaktion wird ein Keton durch eine Persäure zu einem Ester oxidiert. Dabei lagert sich die Persäure an die Carbonylgruppe des Ketons an. Dabei kommt es zu einer Protonenverschiebung. Nach einer weiteren Umlagerung entsteht das Reaktionsprodukt, ein Ester.
Beyer/Walter S. 533
Organikum S.599
Vollhardt S.740
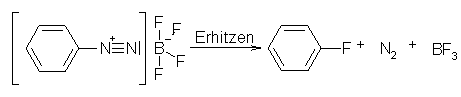
Mit dieser Reaktion kann man Fluorbenzen herstellen, welches mit der Sandmeyer-Reaktion nicht zugänglich ist. Hierbei wird trockenes Benzendiazoniumtetrafluoroborat erhitzt. Dabei entsteht dann Fluorbenzen, Stickstoff und BF3.
Beyer/Walter S.586
Bei dieser Reaktion wird eine Carbonylgruppe an einem nicht aktivierten C-Atom eingeführt. Als Ausgangssubstanz benutzt man ein Alkylnitrit, das in 4-Stellung Wasserstoffatome aufweist, welches mit UV-Strahlung bestrahlt wird. Dabei wird ein Oxim gebildet, welches mit Wasser zu einer Carbonylverbindung hydrolysiert werden kann. Auf diese Weise kann man eine funktionelle Gruppe an einem nicht aktiviertes C-Atom einführen.
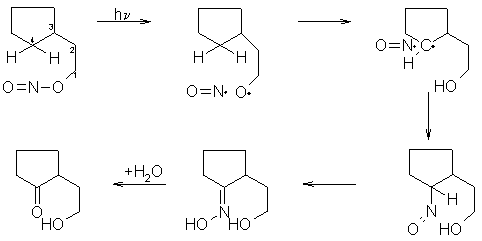
Beyer/Walter S.142
Wirkt ein Säurechlorid, z.B. Phosphor(V)-chlorid, Benzensulfonylchlorid oder eine konzentrierte Mineralsäure auf ein Ketoxim ein, erfolgt eine Umlagerung zu dem entsprechenden Carbonsäureamid oder -anilid. Kommt es zu dieser Umlagerung durch Mineralsäuren, läuft die Umlagerung über die Zwischenstufen: Imminiumion, Oxoniumion, Carbeniumion und ein weiteres Oxoniumion. Diese Umlagerung erfolgt fast immer mit Substituenten, die in anti-Stellung zu einander stehen.
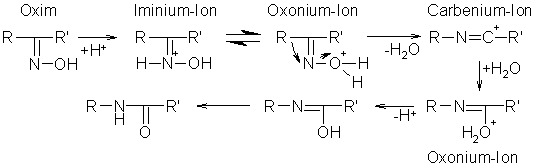
Beyer/Walter S.546
Organikum S.597
Vorlesung
Bei dieser Addition wird aus Benzaldehyd in wäßrigem Ethanol mit Natriumcyanid als Katalysator 2-Hydroxy-1,2-diphenylethanon (Benzoin) gebildet. Primär wird dabei das Cyanidion nucleophil an das positivierte C-Atom der Carbonylgruppe addiert. Es kommt zu einer kationotropen 1,2-Umlagerung („Benzoinumlagerung"), wodurch ein Carbanion gebildet wird. Durch Protonierung wird aus diesem Zwischenprodukt Cyanhydrin gebildet. Durch das Einwirken einer Base wird das Cyanhydrin in der Benzyl-Position zum Benzylanion deprotoniert, was seltener passiert als die Deprotonierung an der Hydroxygruppe (der erste Schritt für die Rückreaktion zum Aldehyd). Das Benzylanion greift nun ein ursprüngliches Aldehyd nucleophil an. Das so erhaltene Anion wird durch das Wasser zum Hydroxycyanhydrin protoniert. Wirkt nun eine Base auf das Hydroxycyanhydrin ein, spaltet es Blausäure (HCN) ab und bildet dadurch das Benzoin.
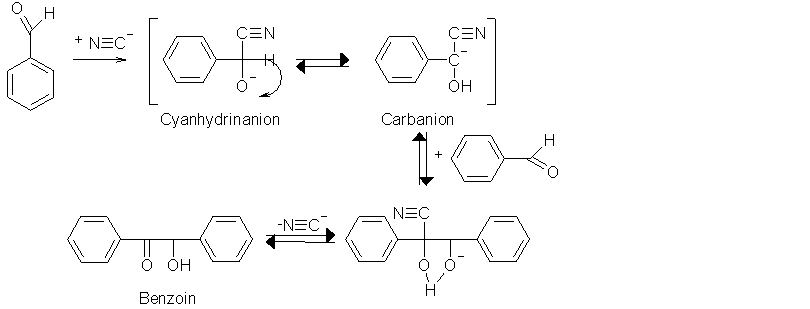 Beyer/Walter
S.535
Beyer/Walter
S.535
Organikum S.474
Vollhard S.1061
Vorlesung
Die Birch-Reduktion ist eine regioselektive Hydrierung von aromatischen Verbindungen in 1,4-Stellung. Als Reagenzien dienen in flüssigem Ammoniak gelöstes Natrium, der Aromat und als Lösungsmittel Ether und ein kurzkettiger Alkohol (Methanol/Ethanol). Die Reaktion beruht auf einer Einelektonenübertragung von dem Metall auf den Aromaten. Dabei wird ein radikalisches Anion gebildet, welches von dem Alkohol zu einem neutralen Radikal protoniert wird. Durch eine weitere Elektronenaufnahme wird aus dem Radikal ein Carbanion, welches erneut von dem Alkohol zu dem Reaktionsprodukt protoniert wird.
Beyer/Walter S.629 und S.723
Vollhardt S.988
Hierbei werden N-acylierte b -Phenylethylamine unter wasserabspaltenden Bedingungen (z.B. Phosphor(V)-oxid, Phosphorchloridoxid [POCl3]) in Benzen oder Tetralin (1,2,3,4-Tetrahydronaphtalin) in ein 3,4-Dihydroisochinolin überführt. Bei dieser Reaktion kommt es zu einer intramolekularen Cyclisierung des Amins. Durch Oxidation mit Kaliumpermanganat oder durch Dehydrierung mit Palladium wird das Dehydroisochinolin in das Isochinolinderivat umgewandelt. Ist das Produkt 1-Methylisochinolin, hat man eine aktivierte Methylgruppe, die mit Aldehyden kondensiert werden können.
Beyer/Walter S.778
Vollhardt S.1161
Die Biuret-Reaktion dient der Prüfung des Harnstoffs auf Reinheit (und zum Nachweis von Peptiden und Proteinen). Erhitzt man Harnstoff auf 150-160°C geht er in Ammoniak und das Aza-Analogon eines Ketens über, welches sofort mit einem weiteren Harnstoffmolekül zu einer Biuretgruppe (H2N-CO-NH-CO-NH2) reagiert. Diese Gruppierung ergibt im Alkalischen mit stark verdünnter Kupfer(II)-sulfatlösung eine rote bis blauviolette Färbung. Diese Reaktion kann auch als Nachweiß von Eiweißen dienen, obwohl in ihnen keine Biuretgruppe auftritt.
Beyer/Walter S.841
Organikum S.425
Vorlesung
Die Blanc-Reaktion stellt eine Einführung einer Chlormethylgruppe (-CH2Cl) in einen aromatischen Kern dar. Bei dieser elektrophilen Reaktion dienen als Ausgangsstoffe ein Aromat, Formaldehyd und Chlorwasserstoff. Als Katalysator dient Zinkchlorid, Aluminiumchlorid oder Phosphorsäure.
Beyer/Walter S.488
Organikum S.346
Nach Bohn-Schmitt lassen sich Polyhydroxyanthrachinone aus einfacheren Hydroxyanthrachinonen herstellen, in dem man sie mit rauchender Schwefelsäure in Gegenwart von Borsäure oxidiert. Die Reaktion kann durch Quecksilber oder Selen katalysiert werden.
Beyer/Walter S.642
Bei dieser Reduktion werden Carbonsäureester zu prim. Alkoholen umgesetzt. Diese Reduktion erfolgt nach Bouveault-Blanc mit Natrium und Ethanol.
Beyer/Walter S.261
Organikum S.449
Bei der von Braun-Reaktion wird Bromcyan (Br-CN) mit tert. Aminen zu quart. Salzen umgesetzt. Aus den Salzen wird Alkylbromid abgespalten, dabei bleibt das eigentliche Reaktionsprodukt, ein Dialkylcyanamid, übrig. Wird das Produkt hydrolysiert, entsteht ein sec. Amin. Daraus ergibt sich die präperative Bedeutung der von Braun-Reaktion, um tert. Amine in sec. Amine umzuwandeln. Als Edukte eignen sich besonders aliphatische Amine. Sollte das tert. Amin mehr als einen Phenylrest tragen, tritt keine Reaktion mit dem Bromcyan ein. Sollte es sich um ein cyclisches tert. Amin handeln, tritt eine Ringöffnung ein.
Beyer/Walter S.365
Die Bucherer-Reaktion überführt Naphtol und seine Derivate in Naphtylamine. Die Reaktion ist nicht geeignet, wenn das Naphtol in m-Stellung zur OH-Gruppe eine Sulfonylgruppe trägt. Hauptsächlich dient diese Reaktion zur Herstellung von a [b ]-Naphtylamin aus dem a [b ]-Naphtol. Als weitere Reagenzien dienen wässerige Ammoniumhydrogensulfit-Lösung und konz. Ammoniak. Das Ammoniumhydrogensulfit addiert sich an das Naphtol, zum Ammoniumsalz der 1[2]-Tetralon-3[4]-sulfonsäure, in einer 3,4-Addition. Unter Hydrogensulfitabspaltung reagiert es zum
a[b]-Naphthylamin.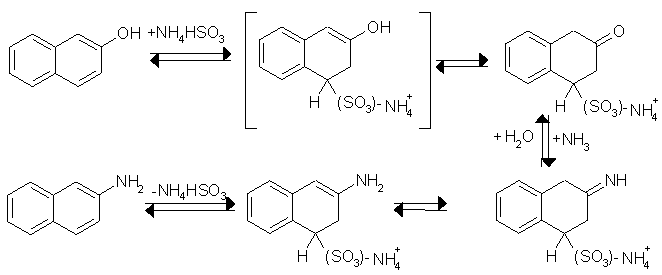 Beyer/Walter
S.633
Beyer/Walter
S.633
Organikum S.464
Bei der Cannizzaro-Reaktion wird ein Aldehyd, welches kein a -ständiges Wasserstoffatom besitzen darf, mit einer Kalilauge (z.B. NaOH) umgesetzt. Dabei wird aus 2 Molen Aldehyd 1Mol prim. Alkohol und 1Mol Alkalisalz der Carbonsäure gebildet. Benutzt man Formaldehyd und einen anderen Aldehyd, so spricht man von einer gekreuzten Cannizzaro-Reaktion. Hierbei entsteht fast nur Natriumformiat und der Alkohol des anderen Aldehyds. Bei der Cannizzaro-Reaktion kommt es zu einer Disproportionierung des Aldehyds in eine Substanz mit niedrigerer Oxidationsstufe (Alkohol) und einer mit höherer Oxidationsstufe (Carbonsäure). Solche Reaktionen bezeichnet man als Oxidoreduktion.
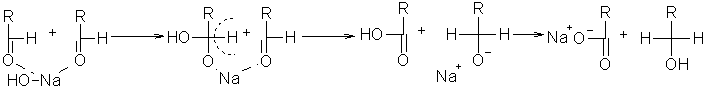 Beyer/Walter
S.201
Beyer/Walter
S.201
Organikum S.507
Vorlesung
Bei der Claisen-Kondensation wird aus 2 Molekülen eines Esters, z.B. Essigsäureethylester (ESEE), ein b -Ketoester, im Fall von ESEE Acetessigester, gebildet. Für diese Reaktion benötigt man als Katalysator ein stark basisches Reagenz, wie z.B. metallisches Natrium, Natriumamid oder Alkoholationen. Die starke Base katalysiert den ersten Schritt der Reaktion, nämlich die Deprotonierung des Esters zum Esterenolat. Das Esterenolat wird jetzt in einer Additions-Eliminierungs-Reaktion mit der Carbonylgruppe eines anderen Estermoleküls zum 3-Ketoester umgesetzt. Die Base deprotoniert den Ketoester unter den gegebenen Bedingungen irreversibel, womit das Gleichgewicht auf die Seite der Produkte verschoben wird. Das Produkt wird wieder erhalten, wenn man das ganze mit wäßriger Säure aufarbeitet. Benutzt man als Base ein Alkoholat, sollte man darauf achten, daß der Ester und das Alkoholat sich vom selben Alkohol ableiten, da es sonst zu einer Umesterung kommen kann. Durch die Claisen-Kondensation lassen sich auch 1,3-Diketone herstellen, wenn man einen Carbonsäureester mit einem Keton, das eine aktivierte Methylgruppe trägt (z.B. Aceton), in Gegenwart eines stark basischen Katalysators (siehe oben) umsetzt.
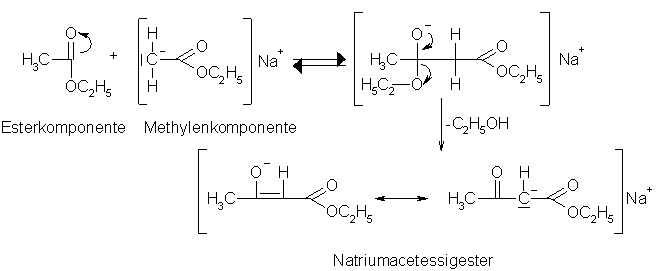 Beyer/Walter
S.296 und S.322
Beyer/Walter
S.296 und S.322
Organikum S.527
Vollhardt S.885 und S.1043
Vorlesung
Bei der Claisen-Umlagerung kann man zwei Arten von Umlagerungen unterscheiden. Zum ersten die Oxo-Claisen-Umlagerung und zum zweiten die Thio-Claisen-Umlagerung. Der Unterschied liegt einfach darin das die Umlagerung zum einen bei Ethern und zum anderen bei Thioethern auftritt. Des weiteren kann man noch zwischen der aliphatischen und der aromatischen Claisen-Umlagerung unterscheiden. Bei der aromatischen Umlagerung können als Edukt Allylphenylether und seine o-substituierten Derivate dienen. Benutzt man Allylphenylether erhält man nach der Umlagerung o-Allylphenol, wobei es zu einer Inversion bei der Allylgruppierung kommt. Hat man als Edukt ein o-substituiertes Derivat, wird ein p-Allylphenolderivat gebildet. Dabei entsteht intermediär ein instabiles Cyclohaxadienonderivat, welches sich durch eine zweite Umlagerung zum p-Allylphenolderivat stabilisiert. Bei dieser Form der Umlagerung kommt es zu einer zweifachen Invertierung. Im aliphatischen Bereich lagern sich Enolether in einer sigmatropen [3,3]-Reaktion zu ungesättigten Aldehyden um. Das ganze geschieht analog zur Cope-Umlagerung.
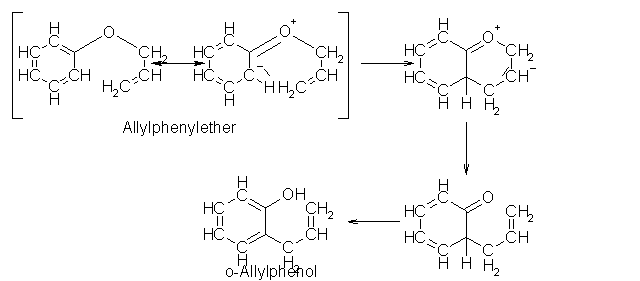 Beyer/Walter
S.500
Beyer/Walter
S.500
Vollhardt S.1009
Bei der Clemmensen-Reduktion werden Ketone in der Hitze durch einwirken von konz. Salzsäure und amalgamiertem Zink bis auf die Methylengruppe (-CH2-) reduziert. Bei Aldehyden gelingt die Reduktion nur in Einzelfällen. Der Reaktionsmechanismus ist noch nicht vollständig geklärt. Es steht nur fest, daß die Reaktion nicht über ein alkoholisches Zwischenprodukt verläuft, da Alkohole unter diesen Bedingungen nicht zu Kohlenwasserstoffen reduzierbar sind.
Beyer/Walter S.217
Organikum S.455
Vollhardt S.680
Vorlesung
Bei dieser Eliminierung wird ein Aminoxid in ein Alken und ein Hydroxylaminderivat gespalten. Die Spaltung wird durch die Positivierung des Stickstoffs in den Aminoxiden erleichtert. Für die Reaktion muß das Aminoxid nicht isoliert werden. Es genügt ein tert. Amin mit Wasserstoffperoxid zu erhitzen. Durch weiteres Erwärmen wird das nun intermediär gebildete Aminoxid in die Produkte gespalten.
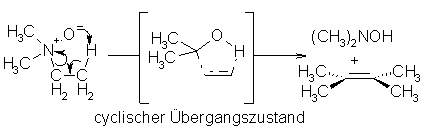
Beyer/Walter S.166
Vollhardt S.954
Vorlesung
Eine Cope-Umlagerung kann bei erhöhter Temperatur bei 1,5-Dienen beobachtet werden. Eine Veränderung kann nur bei unsymmetrischen Dienen beobachtet werden, da sich 1,5-Hexadien zu 1,5-Hexadien isomerisiert. Als einfaches Beispiel nimmt man 3-Methyl-1,5-hexadien, das sich bei ca. 300°C zu 1,5-Heptadien umlagert. Bei der Cope-Umlagerung handelt es sich, wie bei der Claisen-Umlagerung, um eine sigmatrope [3,3]-Reaktion. Die Umlagerung verläuft über einen cyclischen Übergangszustand.
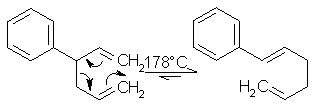
Beyer/Walter S.111
Organikum S.600
Vollhardt S.1010
Bei der Corey-House-Synthese werden Lithiumdialkyl-Cuprate mit Alkylhalogeniden zu Alkanen umgesetzt. Bei dieser Synthese ist es auch möglich Kohlenwasserstoffe mit einer ungeraden Zahl an C-Atomen zu erzeugen.
Beyer/Walter S.57
Die Corey-Seebach-Methode ist eine Möglichkeit um Aldehyde in Ketone zu überführen. Bei der Reaktion wird die Carbonylgruppe umgepolt. Das Ziel der Umpolung ist es einen Reaktanden zu erzeugen, der wie ein Acylanion reagiert. Dazu bedient man sich einer Schwefelverbindung, da Schwefel anionische Zentren besonders gut stabilisieren kann. Dazu wird die Aldehydgruppe mit Propan-1,3-dithiol in ein 1,3-Dithian-Derivat überführt. Das Dithian wird mit n-Butyllithium metalliert. Diese Verbindung stellt ein verkapptes Acylanion dar, welches mit Alkylhalogeniden eine nucleophile Substitution bewirken kann. Das so gewonnene Dithioacetal kann mit Quecksilberchlorid/Quecksilberoxid oder mit Claycop (Bentonit, ein Tonmineral, das mit Kupfer(II)-nitrat imprägniert ist) zum Keton hydrolysiert werden.

Beyer/Walter S.212
Die Criegee-Oxidation dient zur oxidativen Spaltung von Glykolen (Diole mit benachbarten OH-Gruppen). Als Oxidationsmittel dient bei der Reaktion Bleitetraacetat in Eisessig. Die dabei auftretenden Endprodukte sind Aldehyde oder Ketone, je nach eingesetztem Glykol. Des weiteren wird aus dem Bleitetraacetat Bleidiacetat und 2 Teile Essigsäure gebildet.
Beyer/Walter S.303
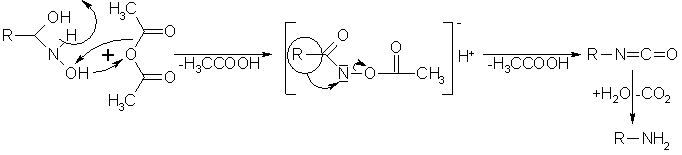
Beim Curtius-Abbau wird ein Carbonsäureazid durch erhitzen in ein prim. Amin umgewandelt. Durch erhitzen von Carbonsäureaziden in alkoholischer Lösung löst sich die Bindung von N2 in der Azidgruppe. Je stärker diese Bindung gelöst wird, desto weiter schreitet die nucleophile Umlagerung der Alkylgruppe fort, und es bildet sich ein Alkylisocyanat. Das Alkylisocyanat addiert sofort ein Molekül Alkohol zu dem entsprechenden Urethan. Durch eine alkalische oder saure Hydrolyse des Urethans erhält man unter Decarboxylierung (-CO2) und Abspaltung des Alkohols das, dem Carbonsäureazid entsprechende, prim. Amin.
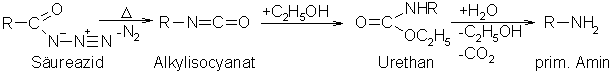
Beyer/Walter S.162
Organikum S.594
Vollhardt S.950
Vorlesung
Auf Grund der besonderen Bindungsverhältnisse bei kleinen Cycloverbindungen (3- und 4-Ringe) kommt es zu Struckturveränderungen. Eine der am häufigsten beobachteten Reaktionen ist dabei die Demjanow-Reaktion. Bei dieser Reaktion wird entweder Cyclobutylamin oder Cyclopropylmethylamin mit salpetriger Säure umgesetzt. In beiden Fällen entstehen als Produkte Cyclobutanol und Cyclopropylmethanol. Das heißt beim Cyclobutylamin kommt es zu einer Ringverengung und beim Cyclopropylmethylamin kommt es zu einer Ringerweiterung.
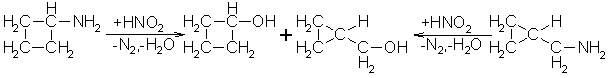
Beyer/Walter S.394
siehe Fischersche-Indolsynthese
Die Dieckmann-Kondensation ist eine Syntheseart für Cycloalkanone, wie z.B. Cyclopentanon. Als Edukte benötigt man einen Diester einer Dicarbonsäure und Natriumethylat bzw. metallisches Natrium in Benzen. Die Dieckmann-Kondensation stellt eine intramolekulare Esterkondensation dar. Der Reaktionsmechanismus ist dem der Claisen-Kondensation ähnlich.
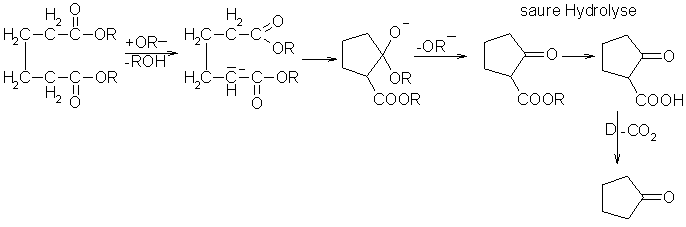 Beyer/Walter
S.397
Beyer/Walter
S.397
Organikum S.489
Vollhardt S.1046
Bei der Diels-Alder-Reaktion reagieren ein Dien und ein Dienophil in einer 1,4-Cycloaddition der aktivierten C=C-Doppelbindung des Dienophils an das konjugierte System des Diens. Dabei entsteht als Produkt ein Derivat des Cyclohexens. Bei dieser Reaktion werden keine Zwischenprodukte beobachtet, d.h. alle Schritte laufen gleichzeitig ab, man spricht von einer konzertierten Reaktion. Das als Dienophil bezeichnete Edukt besitzt eine, durch benachbarte polare Gruppen aktivierte, C=C-Doppelbindung. Als solche polaren Gruppen kommen z.B. in Frage: COOH, CN. Dadurch sind die Dienophile relativ Elektronenarm. Als Beispiele für Dienophile seien genannt: Acrylnitril, Maleinimid, Maleinsäureanhydrid, 1-Nitro-propen, 2-Butenal. Außerdem können auch Ester der, durch die Cº C-Dreifachbindung, relativ „elektronenarmen" Acetylendicarbonsäure dienen. Den Dienen fehlen solche Substituenten, dadurch werden sie relativ elektronenreich. Häufig genutzte Diene sind Butadien; Cyclopentadien; Furan; 1,3-Cyclohexa-dien. Nutzt man die cyclischen Diene, kann es zu 2 verschiedenen Produkten kommen, dem endo-Produkt und dem exo-Produkt. Läßt man die Reaktion bei Raumtemperatur stattfinden wird das endo-Produkt, bei dem das ursprüngliche Dienophil unter dem neu gebildeten 6-Ring liegt, bevorzugt gebildet. Erhöht man die Reaktionstemperatur auf 90°C wird das thermodynamisch günstigere exo-Produkt bevorzugt gebildet.
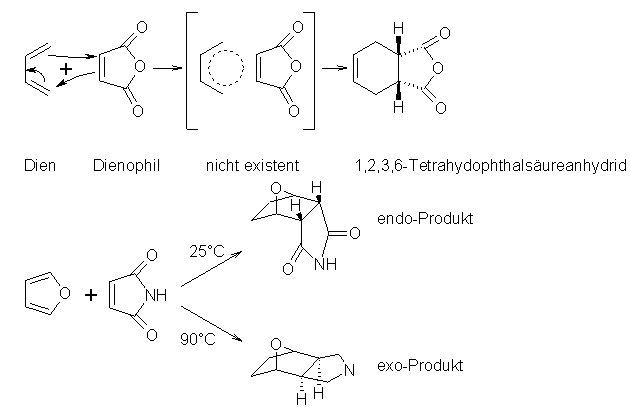
Beyer/Walter S.336
Organikum S.295
Vollhardt S.571
Vorlesung
Doebner-v.Miller-Chinaldinsynthese
Diese Synthese verläuft in Anlehnung an die Skraupsche-Synthese (für Chinolin), mit dem Unterschied, daß als Produkt Chinaldin (2-Methyl-chinolin) gebildet wird. Als Edukte dienen hierbei Anilin und Crotonaldehyd (2-Butenal), welches durch eine Aldolkondensation aus Acetaldehyd in situ hergestellt wird. Die Edukte reagieren in Gegenwart von konzentrierter Salzsäure zu 2-Methyl-1,2-dihydrochinolin. Durch Oxidation dieser Verbindung durch ein Dehydrierungsmittel, wie z.B. Nitrobenzen oder Arsensäure, erhält man das Endprodukt Chinaldin.

Beyer/Walter S.774
Beim Edman-Abbau wird eine Peptidkette schrittweise von der N-terminalen Aminosäure her mit Phenylisothiocyanat abgebaut. Dabei wird aus der Peptidkette und dem Phenylisothiocyanat ein Phenylthioharnstoff-Derivat. Durch Salzsäure wird das Phenylthioharnstoff-Derivat zum entsprechenden Phenylthiohydantonin cyclisiert. Setzt man dieses Produkt einer alkalischen Hydrolyse aus, erhält man die freie Aminosäure. Mit dieser Reaktion kann man Teilsequenzen einer Peptidkette ermitteln.
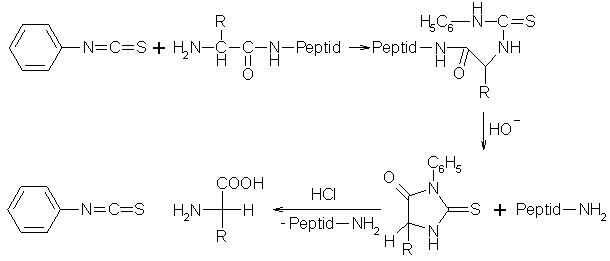
Beyer/Walter S.835
Vollhardt S.1202
Nach dem Einhorn-Verfahren ist es möglich in Alkohole, Phenole und Amine eine Benzoylgruppe einzuführen. Bei dieser Benzoylierung wird der Alkohol mit Benzoylchlorid in Pyridin umgesetzt. Das Pyridin dient als schwache Base nicht nur zum Entziehen des Chlorwasserstoffs, sondern auch als Reaktionsbeschleuniger, da es mit dem Benzoylchlorid einen aktivierten Komplex bildet. Die Reaktion läuft mit bestimmten Derivaten des Pyridins schneller ab als mit Pyridin selber. Als besonders wirkungsvoll hat sich 4-Dimethylaminopyridin (DMAP) erwiesen. Setzt man statt Benzoylchlorid das 3,5-Dinitro-Derivat ein kann man die Reaktion zur Charakterisierung von Alkoholen nutzen, da die Ester dieses Derivats kristallin sind.
Beyer/Walter S.549
Eschweiler-Clarke-Methylierung
Die Eschweiler-Clarke-Methylierung dient der Synthese von tert. Aminen durch eine reduktive Alkylierung von prim. oder sec. Aminen mit Formaldehyd und Ameisensäure. Der Mechanismus ist der Leuckart-Wallach-Reaktion ähnlich.
Beyer/Walter S.229
Vorlesung
Diese Reaktion dient dem Halogenaustausch in halogenierten Kohlenwasserstoffen. Mit ihr kann man z.B. auch Fluoralkane herstellen. Dazu läßt man ein Iodalkan in DMF mit Kaliumfluorid reagieren. Als Zusatz kommt in das DMF noch ein Kronenether und Benzen zur Komplexierung des Kaliums. Dabei entsteht ein Fluoralkan und Kaliumiodid.
Organikum S.220
Vorlesung
Mit der Fischerschen-Indolsynthese kann man Indol und seine Derivate synthetisieren. Als Edukte werden ein Phenylhydrazon einer Ketovebindung und Zinkchlorid (Schwefelsäure/Borfluorid) als Kondensationsmittel verwendet. Das Phenylhydrazon reagiert bei Anwesenheit von Zinkchlorid unter Ringkondensation zu dem entsprechenden Indol-Derivat. Das Phenylhydrazon wird aus einem Phenylhydrazin und einem Keton unter Wasserabspaltung synthetisiert. Durch eine Diaza-Cope-Umlagerung verschiebt sich die C=N-Doppelbindung zu einer C=C-Doppelbindung. Nach Ammoniak Abspaltung kommt es bei Anwesenheit des Kondensationsmittels zum Ringschluß des Indolrings. Verwendet man Acetaldehydphenylhydrazon als Edukt wird Indol als Produkt gebildet.
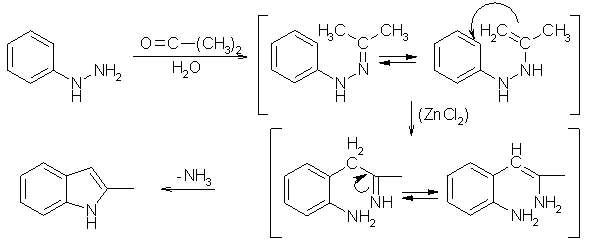
Beyer/Walter S.725
Organikum S.601
Vollhardt S.1149
Vorlesung
Die Fischer-Tropsch-Synthese ist eine Möglichkeit Kraftstoffe ohne Erdöl herzustellen. Man benutzt dazu Synthesegas, welches mit Wasserstoff angereichert ist.
Beyer/Walter S.90
Vollhardt S.260
Das Frank-Caro-Verfahren dient zur Herstellung von Calciumcyanid, welches zur Herstellung von Cyanamid benötigt wird. Dazu wird Calciumcarbid im Stickstoffstrom auf 1000°C erhitzt.
Beyer/Walter S.368
Die Friedel-Crafts-Acylierung ist eine elektrophile Substitution an einem aromatischen Kern. Als Edukte wird ein aromatischer Kohlenwasserstoff (er kann auch Heteroatome tragen), ein aliphatisches oder aromatisches Säurechlorid oder Säureanhydrid und eine Lewissäure, z.B. Aluminiumchlorid. Das Säurechlorid reagiert mit der Lewissäure in einem Gleichgewicht zu einem Komplex, aus der Lewissäure und dem Säurechlorid, und zu einem Acylkation. Das Acylkation greift den aromatischen Kern nun unter Bildung eines nicht aromatischen Carbeniumions. Durch Deprotonierung wird das aromatische System zurückgebildet. Das so entstandene Keton hat die Lewissäure noch komplex gebunden. Zur Überführung in das Keton muß anschließend noch hydrolysiert werden. Im Gegensatz zur Friedel-Crafts-Alkylierung, kommt es bei der Acylierung nicht zu einer 2. Substitution, da die CO-Gruppe, als Substituent 2. Ordnung, einen Eintritt weiterer Ketogruppen erschwert.
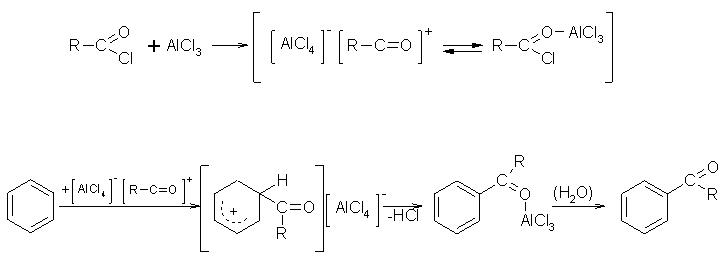 Beyer/Walter S.541
Beyer/Walter S.541
Organikum S.339
Vorlesung
Diese Reaktion führt zu alkylierten aromatischen Kohlenwasserstoffen. Als Ausgangsstoffe werden hierbei Alkylhalogenide, z.B. Ethylchlorid, aromatische Kohlenwasserstoffe, z.B. Benzen, und katalytische Mengen Aluminiumchlorid. Aluminiumchlorid besitzt, wie jede Lewissäure, nur ein Elektronensextett und hat daher die Bestrebung das Halogenatom des Alkylhalogenids komplex zu binden. Dabei muß es nicht zu einer vollständigen Trennung der Halogen-Kohlenstoff-Bindung kommen. Das sehr stark positivierte C-Atom greift nun elektrophil am
p-Elektronensextett des Aromaten an. Dabei bildet sich ein primärer p-Komplex, der sich zu einem Carbeniumion, auch s-Komplex genannt, stabilisiert. Die Ladung des Ions ist dabei vollständig delokalisiert. Der Reaktionsmechanismus erinnert dabei stark an die elektrophile Addition an eine C=C-Doppelbindung. Sie unterscheidet sich jedoch in der weiteren Reaktion. Am Alken addiert sich ein Anion, der s-Komplex spaltet über einen sekundären p-Komplex ein Proton ab und geht dabei in den energetisch begünstigten aromatischen Zustand über. Dabei kommt es zu heftiger Halogenwasserstoffentwicklung. Ein großer Nachteil der Friedel-Crafts-Alkylierung ist, das sie nicht auf der primären Stufe stehen bleibt, da die Alkylierungsprodukte meist schneller mit dem Alkylhalogenid reagiert als das Benzen. Da diese Reaktion reversibel ist, kann es zu einer Isomerisierung, sowohl intra- als auch intermolekular kommen. Darum ist das entstehende Produkt immer thermodynamisch gesteuert. Statt Aluminiumchlorid wird zuweilen auch Eisen(III)-chlorid, Zinn(IV)-chlorid, Zinkchlorid, Borfluorid, wasserfreie Flußsäure, konzentrierte Schwefelsäure oder Phosphorsäure als Katalysator verwendet.
Beyer/Walter S.477
Organikum S.335
Vollhardt S.643
Vorlesung
Die Friedländer-Synthese dient zur Herstellung von Chinolin aus o-Aminobenzaldehyd und Aldehyden oder Ketonen. Die Aldehyde und Ketone müssen eine aktivierte CH2-Gruppe in Nachbarstellung zur Ketogruppe tragen. Unter alkalischen Bedingungen kommt es zu einer Ringkondensation zum Chinolin oder einem seiner Derivate.
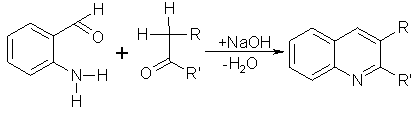
Beyer/Walter S.774
Vollhardt S.1161
Erhitzt man Phenylester einer aliphatischen oder aromatischen Carbonsäure mit Aluminiumchlorid, Borfluorid, Zinkchlorid oder Titan(IV)-chlorid in wasserfreiem Nitrobenzen, kommt es zur Fries-Umlagerung in o- der p-Acylphenole. Teilweise ist es eine intramolekulare Umlagerung. Das o-Produkt bildet sich hauptsächlich bei Temperaturen von über 100°C, wohingegen das p-Produkt in der Kälte bevorzugt gebildet wird.
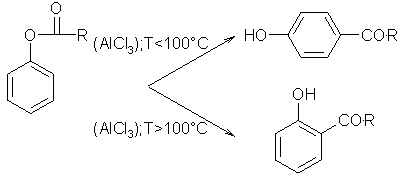
Beyer/Walter S.500
Organikum S.585
Die Gabriel-Synthese ist eine Möglichkeit um prim. Amine herzustellen. Als Edukte werden für diese Synthese ein Alkylhalogenid, in der Regel ein Alkylchlorid, und Phthalimidkalium verwendet. Erhitzt man das Imid und das Alkylhalogenid bildet sich zu erst ein N-Alkylphthalimid. Das N-Alkylphthalimid spaltet sich durch Hydrazin in das prim. Amin und in Phthalhydrazid.
 Beyer/Walter
S.163
Beyer/Walter
S.163
Organikum S.213
Vollhardt S.946
Vorlesung
Die Gattermann-Aldehydsynthese ist eine Sonderform der Gattermann-Koch-Synthese um aus Phenolen und Phenolethern das entsprechende Aldehyd zu synthetisieren, da dies durch die Gattermann-Koch-Synthese nicht möglich ist. Bei der Gattermann-Aldehydsynthese dienen Phenol, dessen Derivate oder Phenolether, wasserfreie Blausäure, trockenen Chlorwasserstoff und als Katalysator Aluminiumchlorid oder das schwächere Zinkchlorid als Ausgangssubstanzen. Benutzt man mehrwertige Phenole kann man die Reaktion ohne den Katalysator durchführen. Als Lösungsmittel kann man Ether, Chloroform, Benzen, Chlorbenzen oder 1,1,2,2-Tetrachlorethan benutzen. Die Blausäure und der Chlorwasserstoff bilden das reaktive, nicht isolierbare Teilchen Formimidchlorid. Das Formimidchlorid reagiert mit dem aromatischen Alkohol oder Ether zum Aldiminhydrochlorid, welches sich beim erwärmen in wäßriger Säure zum Aldehyd hydrolysiert. Bei dieser Reaktion entsteht vornehmlich ein zur Hydroxygruppe bzw. Ethergruppe, in p-Stellung formyliertes Endprodukt. Die Reaktion ist ein Spezialfall der Friedel-Crafts-Acylierung. Diese Synthese wird heute aber nicht mehr so häufig angewandt, da die Ausbeuten bei der Vilsmeier-Haack-Synthese erheblich höher sind.
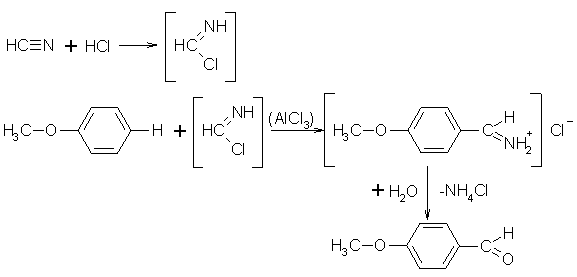
Beyer/Walter S.537
Organikum S.343
Vorlesung
Die Gattermann-Koch-Synthese überführt aromatische Kohlenwasserstoffe in das entsprechende Aldehyd. Für die Synthese verwendet man trockenen Chlorwasserstoff, Kohlenmonoxid, wasserfreien Aromaten, Aluminiumchlorid und Kupfer(I)-chlorid. Der Chlorwasserstoff und der Kohlenmonoxid reagieren bei Anwesenheit von Aluminiumchlorid und Kupfer(I)-chlorid wie das instabile Formylchlorid. Das Formylchlorid reagiert mit dem Aromaten zu einem Benzaldehyd-Derivat, bei dem die Aldehydgruppe meistens in p-Stellung zum vorhandenen Substituenten eintritt. Auch die Gattermann-Koch-Synthese ist ein Spezialfall der Friedel-Crafts-Acylierung. Auch sie wurde durch andere Reaktionen, wegen der stark schwankenden Ausbeuten, verdrängt.
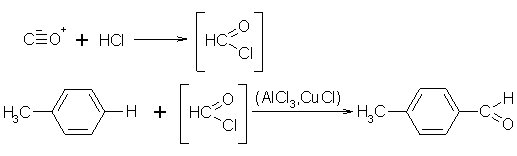
Beyer/Walter S.537
Organikum S.343
Vorlesung
Mit der Gomberg-Bachmann-Reaktion erreicht man eine Arylierung am Benzenkern von Diazoniumsalzen. Um dies zu erreichen, muß man die Diazoniumsalzlösung mit einem Aromaten, z.B. Benzen, unter Rühren innig vermischen. Nun muß man verdünnte Alkalilauge, z.B. Natronlauge, hinzu tropfen. Verwendet man Benzendiazoniumchlorid als Diazoniumsalz, entsteht dabei mit etwa 20% Ausbeute Biphenyl. Die Reaktion verläuft über das Diazohydroxid und reagiert nach einem Radikalmechanismus weiter. Nach Deprotonierung bildet sich aus dem Radikal das Biphenyl.
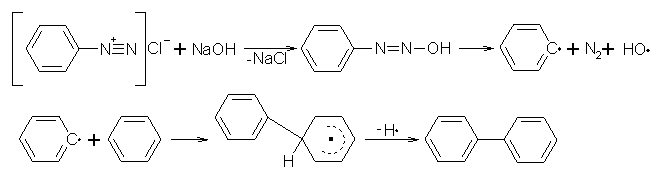 Beyer/Walter
S.586
Beyer/Walter
S.586
Organikum S.566
Die Graebe-Ullmann-Synthese stellt eine Reaktion dar, durch die Carbazol (Dibenzopyrrol) zugänglich ist. Dazu wird o-Aminodiphenylamin mit salpetriger Säure zur Reaktion gebracht. Es reagiert unter Wasserabspaltung und Diazotierung und anschließender intramolekularer Kupplung zum 1-Phenyl-1,2,3-benzotriazol, das beim Erhitzen Stickstoff abspaltet und nahezu quantitativ in Carbazol übergeht.
Beyer/Walter S.734
Die Grignard-Reaktion kann zu prim., sec., tert. Alkoholen und zu Carbonsäuren führen. Als Edukte für alle 4 Produkte dienen sogenannte Grignard-Verbindungen. Für prim. Alkohole kommt als 2. Edukt nur Formaldehyd in Frage. Möchte man einen sec. Alkohol herstellen kann man alle anderen Aldehyde verwenden, für tert. Alkohole sind Ketone einzusetzen. Um Carbonsäuren zu produzieren leitet man in die etherische Grignard-Lösung trockenes Kohlendioxid ein.
Eine Grignard-Verbindung ist ein Alkyl- oder Arylmagnesiumhalogenid, meist ein Bromid. Um es herzustellen löst man Magnesium in absolut trockenem Ether und gibt ein Alkyl- oder Arylhalogenid hinzu. Dabei bildet sich unter Umpolung am C-Atom das Alkyl- oder Arylmagnesiumhalogenid. Die Grignard-Verbindung hat 2 Moleküle Ether komplex am Magnesium gebunden. Es wird vermutet, daß die Verbindung in Lösung vorwiegend dimolekular vorliegt, der Einfachheit wegen läßt man in der Regel den Ether und die Dimolekularität beim schreiben weg. Es wird nur R-MgX, wobei R dem organischen Rest und X dem Halogenid entspricht, geschrieben. Grignard-Verbindungen sind extrem Wasser empfindlich und reagieren heftig zum entsprechenden Kohlenwasserstoff und Magnesiumhydroxyhalogenid, darum muß man im absolut wasserfreiem Medium arbeiten. Der Magnesiumhalogenidrest lagert sich immer an das elektronegativste Element des 2. Eduktes als Kation an. Der organische Rest reagiert als Carbanion mit dem benachbarten C-Atom. Hydrolysiert man das so gewonnene Zwischenprodukt wird der Magnesiumhalogenidrest durch ein Proton ausgetauscht und es entsteht je nach Edukt das entsprechende Produkt und Magnesiumhydroxyhalogenid.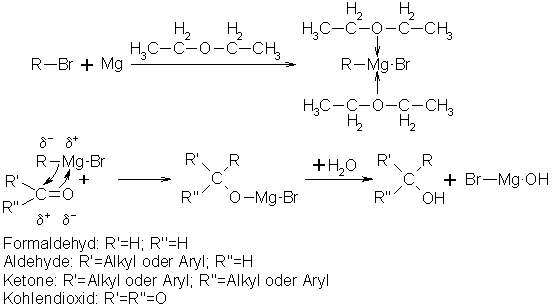
Beyer/Walter S.187
Organikum S.515
Vollhardt S.270
Vorlesung
Hantzsche-Dihydropyridinsynthese
Bei der Hantzschen-Dihydropyridinsynthese werden ein
b-Ketosäureester, ein Aldehyd und Ammoniak im Verhältnis 2:1:1 miteinander zur Reaktion gebracht. Dabei gehen sowohl der b-Ketosäureester und das Aldehyd, als auch der b-Ketosäureester und der Ammoniak eine Art Knoevenagel-Reaktion ein. Dabei wird mit dem Ester ein ungesättigter Ketosäureester und dem Ammoniak wird ein Enamin gebildet. Die beiden ersten Reaktionsprodukte reagieren in einer Art Michael-Addition weiter zu einem Dihydropyridin oder einem seiner Derivate.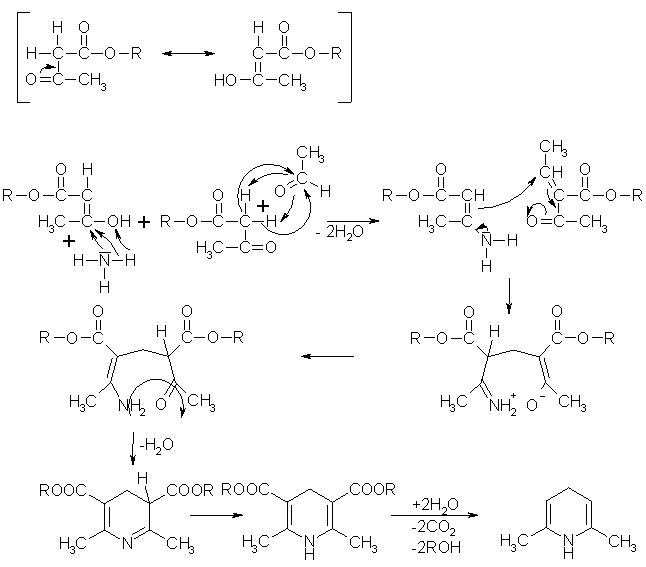 Beyer/Walter
S.767
Beyer/Walter
S.767
Organikum S.531
Vollhardt S.1153
Vorlesung
Diese Synthese ist eine Kondensation von
a-Halogenketoverbindungen mit Thioamiden verschiedener Art. Durch diese Kondensation entstehen Thioazolderivate. Der erste Schritt dieser Reaktion ist die nucleophile Substitution des Halogenatoms durch den Schwefel. Der nächste Schritt stellt die Ringkondensation dar, welche eine Gleichgewichtsreaktion ist. Nach dem Ringschluß spaltet das Produkt Wasser ab. Durch entziehen des Wassers kann das Gleichgewicht auf die Seite des Thiazolderivats verschoben werden.Beyer/Walter S.749
Organikum S.411
Durch diese Synthese ist Phenanthren aus Naphthalin zugänglich. Als ersten Reaktionsschritt führt man eine Friedel-Crafts-Acylierung mit Naphthalin und Bernsteinsäureanhydrid durch. Daraus ergibt sich als erstes Reaktionsprodukt
b-[1-Naphthoyl]-propionsäure. b-[1-Naphthoyl]-propionsäure wird durch eine Clemmensen-Reduktion mit amalgamiertem Zink und Salzsäure in die g-[1-Naphthyl]-buttersäure überführt. Der g-[1-Naphthyl]-buttersäure wird mit konzentrierter Schwefelsäure unter Ringschluß in der 2-Stellung Wasser entzogen. Das dabei entstehende Keton wird durch eine weitere Clemmensen-Reduktion und anschließender Dehydrierung durch Selen in Phenanthren überführt. Ein Ringschluß in 1-Stellung, der zu Anthracen führen würde, ist unter diesen Bedingungen nicht möglich. Die Haworth-Synthese wird hauptsächlich zur Synthese der Alkylderivate des Phenanthrens verwendet.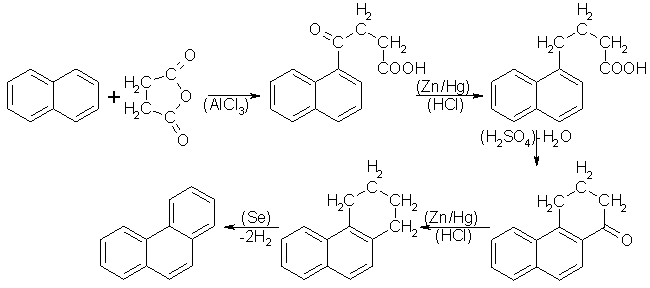 Beyer/Walter
S.645
Beyer/Walter
S.645
Hell-Volhard-Zelinsky-Reaktion
Die Hell-Volhard-Zelinsky-Reaktion wird ausschließlich zur Synthese von
a-Bromcarbonsäuren verwendet. Dazu wird die zu halogenierende Carbonsäure mit rotem Phosphor versetzt. Zu diesem Substrat wird langsam Brom zu getropft, dann wird auf 80°C erwärmt. Es bildet sich intermediär Phosphor(III)-bromid, welches mit der Carbonsäure zuerst zum Säurebromid reagiert. Das Säurebromid geht dann leicht in das entsprechende a-Bromcarbonsäure-bromid über. Das a-Bromcarbonsäurebromid reagiert mit einem weiteren Molekül Carbonsäure zu einem Molekül Säurebromid und einem Molekül a-Bromcarbonsäure. Darum genügen meist kleinere Mengen Phosphor.
Beyer/Walter S.272
Vollhardt S.841
Vorlesung
Es gibt 2 verschiedene Synthesen nach Heumann. Bei der ersten Heumanschen-Indigosynthese wird Anilin mit Chloressigsäure unter Chlorwasserstoffentwicklung zu Phenylglycin kondensiert. Das Phenylglycin wird bei 180-200°C mit Natriumamid einer alkalischen Schmelze unterzogen. Dabei entsteht unter Abspaltung von Wasser und Ringschluß ein kondensiertes Ringsystem mit dem Namen Indoxyl. Indoxyl reagiert mit Luftsauerstoff zu Indigblau (Indigo). Der 2. Syntheseweg geht von, der aus Anthranil- und Chloressigsäure zugänglichen, Phenylglycin-o-carbonsäure aus. Schmilzt man Phenylglycin-o-carbonsäure mit Alkali entsteht in quantitativer Ausbeute 2-Indoxycarbonsäure, auch 3-Oxoindolinon-2-carbonsäure genannt. Erwärmt man 2-Indoxycarbonsäure reagiert sie unter Decarboxylierung zu Indoxyl. Indoxyl reagiert mit Luftsauerstoff zu Indigblau.

Beyer/Walter S.729
Die Hinsberg-Reaktion dient der Unterscheidung und Trennung von prim., sec. und tert. Aminen mit Benzensulfonylchlorid. Mit Benzensulfonylchlorid bilden nur prim. und sec. Amine kristalline Benzensulfonamide, tert. Amine bleiben in der Lösung. Die Benzensulfonamide der prim. und sec. Amine können durch behandeln mit Alkali getrennt werden, da die Benzensulfonamide der prim. Amine in den Kalilaugen löslich sind, die der sec. Amine dagegen unlöslich.
Beyer/Walter S.161
In etherischer Lösung können mit der Hoesch-Ketonsynthese aus mehrwertigen Phenolen und Säurenitrilen in Gegenwart von Chlorwasserstoff aromatische Ketone produziert werden. Im ersten Schritt reagieren Chlorwasserstoff und Säurenitril zu einem Säureimidchlorid, welches im nächsten Schritt mit dem Phenol zu einem Ketiminhydrochlorid, das nur intermediär auftritt. Die Verseifung mit Wasser führt zu dem aromatischen Keton und zu Ammoniumchlorid.
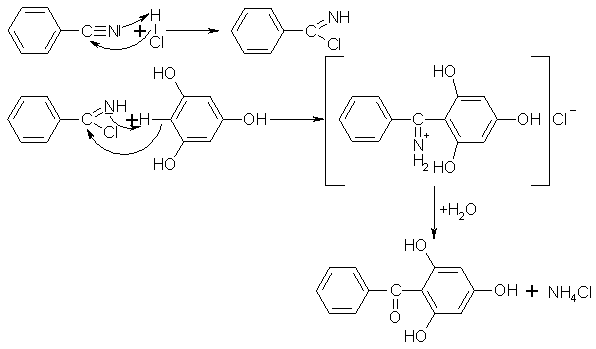
Beyer/Walter S.542
Organikum S.343
Beim Hofmann-Abbau wird ein Carbonsäureamid unter Kettenverkürzung von einem C-Atom zu einem prim. Amin abgebaut. Für den Abbau wird das Carbonsäureamid in einem geringen Überschuß einer wäßrigen Natriumhypobromit-Lösung , die aus Natronlauge und Brom zugänglich ist, gelöst und schnell auf ca. 70°C erhitzt. Dabei bildet sich zuerst das N-Bromamid der Carbonsäure. Das Amid reagiert mit Natronlauge unter Deprotonierung, anschließendem Abspalten des Bromid-Ions und Umlagerung zum Alkylisocyanat. Wird das Isocyanat hydrolysiert geht es unter Abspaltung von Kohlendioxid in das prim. Amin über.

Beyer/Walter S.162
Organikum S.593
Vollhardt S.892
Vorlesung
Unter einer Hofmann-Alkylierung versteht man das Umsetzen eines Alkylhalogenids mit Ammoniak. Dabei entsteht ein Gemisch aus prim., sec., tert. Aminen und quart. Ammoniumsalzen.
Beyer/Walter S.161
Vorlesung
Durch die Hofmann-Eliminierung gehen höhere Tetraalkylammoniumhydroxide in ein tert. Amin, ein Alken und Wasser über. Ist in dem Tetraalkylammoniumhydroxid eine Ethylengruppe enthalten, tritt immer Ethen als Spaltprodukt auf, dieser Fall wird
b-Eliminierung genannt. Sollte die quart. Ammoniumbase Substituenten enthalten die mehrere isomere Olefine bilden können, wird gemäß der Hofmann-Regel, das Alken mit der geringsten Zahl an Alkylgruppen gebildet. Kommt es bei Alkylhalogeniden zu einer b-Eliminierung wird nach der Saytzew-Regel überwiegend das am stärksten verzweigte Isomer gebildet.
Beyer/Walter S.160
Organikum S.251
Vollhardt S.951
Vorlesung
Durch diese Reaktion wird eine Möglichkeit zur Olefinierung von Carbonylverbindungen aufgezeigt. Als Edukte können dabei Phosphonsäureester und Phospinoxide, die eine
a-ständige, acide CH-Gruppe tragen, verwendet werden. Das Edukt wird dann mit Natriumhydrid (NaH) in 1,2-Dimethoxyethan als Lösungsmittel zum Olefin (Alken) umgesetzt.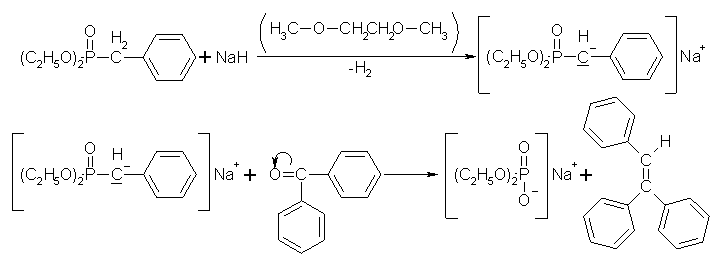 Beyer/Walter
S.178
Beyer/Walter
S.178
Das Houndry-Verfahren ist das katalytische cracken von den hochsiedenden Erdölfraktionen zu Benzinen.
Beyer/Walter S.88
Diese Methode ist eine modifizierte Wolff-Kishner-Reduktion. Mit dieser Reaktion wird eine CO-Gruppe in eine CH2-Gruppe überführt. Im Gegensatz zur Wolff-Kishner-Reduktion muß das Ketohydrazon nicht isoliert werden. Die Carbonylverbindung wird bei dieser Modifikation in Ethylenglykol mit hochprozentigem Hydrazinhydrat und Natriumhydroxid auf ca. 150°C erhitzt. Eine weitere Möglichkeit ist die Reduktion in Dimethylsulfoxid (DMSO) mit Kalium-tert.-butylat.
Beyer/Walter S.217
Die Hunsdiecker-Reaktion dient zur Herstellung von Alkylhalogeniden aus den Silbersalzen von Carbonsäuren in Tetrachlorkohlenstoff mit Brom oder Iod. Dabei decarboxyliert das Salz der Säure unter Bildung des Alkylhalogenids.
Beyer/Walter S.127
Vollhardt S.844
Vorlesung
Bei der Iwanow-Reaktion wird Phenylessigsäure zu einer
b-Hydroycarbonsäure umgesetzt. Dazu wird die Phenylessigsäure, deren zur Carboxylgruppe a-ständiges Wasserstoffatom sauer reagiert, mit einem Grignard-Reagenz, z.B. Isopropylmagnesiumchlorid, in etherischer Lösung umgesetzt. Dabei entsteht ein Dianion, da auch das zur Carboxylgruppe a-ständige Wasserstoffatom abgespalten wird. Gibt man zu der Grignard-Lösung nun Aldehyde oder unsymmetrische Ketone, erhält man nach saurer Hydrolyse substituierte b-Hydroycarbonsäuren. Es lassen sich dabei meist 2 diastereomere Formen der Säure isolieren. Die Addition verläuft ähnlich der Hydroxycarbonsäurensynthese mit der Reformatsky-Reaktion.Beyer/Walter S.555
Bei der Knoevenagel-Reaktion bringt man Anionen von
b-Dicarbonylverbindungen oder ähnlichen Verbindungen mit aktivierter Methylengruppe (Methylenkomponente) und ein Aldehyd oder Ketone, die Carbonylkomponente, zur Reaktion. Die Anionen der Dicarbonylverbindungen erhält man, wenn die b-Dicarbonylverbindung mit katalytischen Mengen einer schwachen Aminbase, wie z.B. Diethylamin, behandelt wird. In Gegenwart des Aldehyds oder Ketons reagiert das Carbanion mit einer nucleophilen Addition an das positivierte C-Atom der Carbonylgruppe und anschließender Wasserabspaltung zum ungesättigten Dicarbonsäurediester. Benutzt man a,b-ungesättigte Carbonylverbindungen, reagieren diese mit einer 1,4-Addition, der Michael-Addition.
Beyer/Walter S.328
Organikum S.476
Vollhardt S.1054
Vorlesung
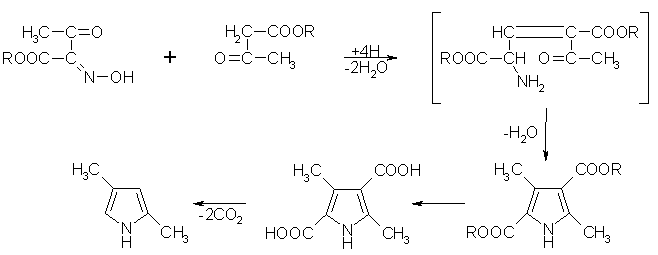
Mit dieser Synthese sind substituierte Pyrrole zugänglich. Als Edukte werden hierbei
a-Aminoketoverbindungen und Carbonylverbindungen mit einer aktivierten Methylengruppe, z.B. Acetessigester, verwendet. Der a-Aminoketon wird meist intermediär erzeugt, z.B. aus Hydroxyiminoacetessigester. Dazu wird diese Verbindung mit Zinkstaub und Eisessig reduziert. Das Reduktionsprodukt reagiert mit dem Acetessigester sofort zu 3,5-Dimethyl-pyrrol-2,4-bis(carbonsäureethylester) weiter. Nach der Cyclokondensation wird der Ester durch alkalische Hydrolyse und Decarboxylierung in das 2,4-Dimethylpyrrol überführt. Beyer/Walter
S.707
Beyer/Walter
S.707
Organikum S.480
Durch die Koch-Haaf-Reaktion werden tert. Carbonsäuren technisch Hergestellt. Dazu verwendet man ein Alken, z.B. 2-Methylpropen, und setzt es mit konzentrierter Schwefelsäure und Kohlenmonoxid um. Nach anschließender Hydrolyse erhält man eine tert. Carbonsäure.
Beyer/Walter S.232
Die Kolbe-Nitrilsynthese ist eine Cyanid-Alkylierung. Dabei wird ein Alkylhalogenid in wäßrig, alkoholischer Lösung mit Kaliumcyanid umgesetzt. Dabei entsteht als Hauptprodukt das Alkylnitril, nur in geringen Mengen das Alkylisocyanid. Das liegt daran, daß die nach einem SN2-Mechanismus ablaufende Reaktion bevorzugt am nucleophileren Atom, dem C-Atom, stattfindet. Das Isocyanid wird durch schütteln mit verdünnter Salzsäure entfernt. Die Reaktionsfähigkeit der Alkylhalogenide nimmt in der Reihenfolge Iodid, Bromid und Chlorid ab.
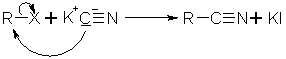
Beyer/Walter S.268
Organikum S.223
Vorlesung
Mit der Kolbe-Schmitt-Synthese wurde ein Weg gefunden um Salicylsäure herzustellen. Die Synthese findet bei 120-140°C und einem Druck von 0,5-0,6 MPa (5-6 bar) statt. Dazu läßt man unter diesen Bedingungen Kohlendioxid auf trockenes Natriumphenolat einwirken. Dabei findet eine direkte Carboxylierung am Phenolation statt, welche eine elektrophile Substitution in o-Stellung zum Natriumsalicylat darstellt. Verwendet man statt Natriumphenolat das Kaliumphenolat findet keine o-, sondern eine p-Substitution statt. Es wird p-Hydroxybenzoesäure gebildet.
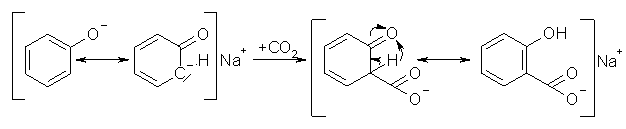
Beyer/Walter S.552
Vollhardt S.1009
Bei der Kolbe-Synthese handelt es sich um eine anodische Oxidation von Carbonsäuren. Bei der Synthese kommt es zu 2 Einelektronenübertragungen, bei der das sehr instabile RCOO× -Radikal gebildet wird, das sich unter Decarboxylierung stabilisiert. Es entsteht dabei ein Alkylradikal, das sich mit einem weiteren Alkylradikal dimerisiert. Dabei werden nur Alkane mit einer geraden Anzahl von C-Atomen gebildet.

Vollhardt S.843
Vorlesung
Eine Kupplungsreaktion ist eine Verknüpfung einer aromatischen Diazoverbindung mit einer zweiten Komponente. Die zweite Komponente kann ein aromatisches Amin oder ein Phenol sein. Bei Kupplungen unterscheidet man N-Kupplungen und C-Kupplungen.
Bei dieser Kupplungsreaktion können sowohl Phenole, als auch tert. Amine als erstes Edukt dienen. Als zweite Ausgangssubstanz wird genau wie bei der N-Kupplung ein aromatisches Diazoniumion verwendet. In schwach saurer Lösung reagieren die Edukte zu p-Aminoazo-(bzw. Hydroxyazo)verbindungen.
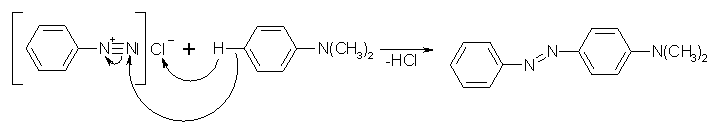
Beyer/Walter S.589
Vorlesung
Bei der N-Kupplung dienen ein aromatisches Diazoniumion und ein prim. Amin als Ausgangssubstanz. Die Reaktion verläuft in einem schwach sauren Milieu zu einer Diazoaminoverbindung. Versetzt man diese Verbindung mit dem Aminhydrochlorid und erwärmt auf ca. 40°C, lagert sich die Diazoaminoverbindung intermolekular in eine Aminoazoverbindung um. Diese Umlagerung erfolgt über eine C-Kupplung.
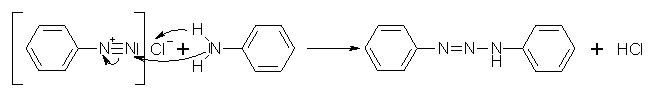 Beyer/Walter
S.588
Beyer/Walter
S.588
Vorlesung
Mit dieser Reaktion wird eine reduktive Carbonylaminierung durchgeführt, z.B. mit Acetophenon und Ammoniumformiat, wobei als Produkt
a-Phenylethylamin entsteht. Als Reduktionsmittel dient in diesem Fall die entstehende Ameisensäure. Dabei werden, im Gegensatz zur Reduktion mit Natrium, am Benzenkern haftende Halogenatome oder Nitrogruppen nicht angegriffen.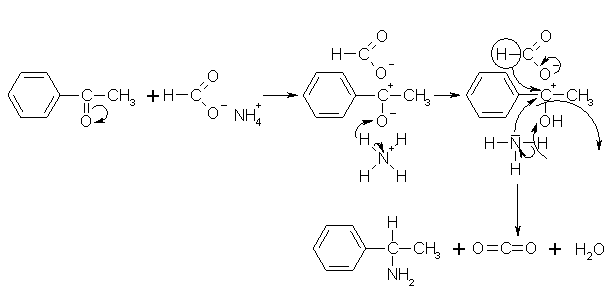
Beyer/Walter S.526
Organikum S.509
Vorlesung
Lobry de Bruyn-vanEkenstein-Umlagerung
Die Lobry de Bruyn-vanEkenstein-Umlagerung stellt das Gleichgewicht zwischen Glycerolaldehyd und Dihydroxyaceton dar. Dabei kommt es in wasserfreiem Pyridin unter Protonenwanderung über ein Endiol zu einer Umlagerung.
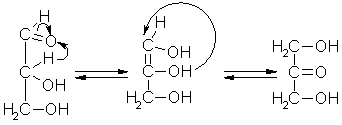
Beyer/Walter S.318 und S.435
Durch thermische Dehydratisierung in indifferenten Lösungsmitteln (Ether, Benzen) von Hydroxamsäuren, oder besser in Gegenwart von Phosphor(V)-oxid oder Essigsäureanhydrid, kommt es zum Lossen-Abbau. Dabei kommt es zu einer Bildung einer Acetylverbindung, die sich nucleophil zum Alkylisocyanat umlagert. Wird das Alkylisocyanat hydrolysiert, bildet sich ein prim. Amin.
 Beyer/Walter
S.162
Beyer/Walter
S.162
Die Madelung-Synthese ist eine weitere Möglichkeit zur Indolproduktion. Als Edukte dienen hierbei Kalium-tert.-butylat, als Kondensationsmittel, und 2’-Methylformanilid. Die beiden Edukte werden in einer Stickstoffatmosphäre auf 350-360°C erhitzt, dabei kommt es zu einem intramolekularem Ringschluß. Durch diese Reaktion entsteht Indol in 79%-Ausbeute.
Beyer/Walter S.725
Die Mannich-Reaktion ist eine Kondensation von einer CH-aciden Verbindung, die Methylenkomponente, mit Formaldehyd bei Gegenwart von sec. oder prim. Aminen, der Aminkomponente, die häufig als Hydrochloride vorliegen. Setzt man Ketone mit Formaldehyd unter Zugabe einer äquimolaren Menge Ammoniumsalz um, erfolgt eine Kondensation zum
b-Aminoketon. Es ist eine a-Aminomethylierung erfolgt, bei der das aktive Wasserstoffatom des Ketons durch eine Dialkyl- bzw. Monoalkylaminomethylgruppe ersetzt wurde. Der Reaktionsmechanismus im neutralen oder saureren Milieu führt zunächst, durch Addition des einsamen Elektronenpaars des Amin-Stickstoffs an das positivierte C-Atom der Carbonylgruppe des Formaldehyds, zu einem Addukt, das zunächst protoniert wird und unter Wasserabspaltung zu einem Methyleniminiumkation weiter reagiert. Das Methyleniminiumkation ist durch die Mesomerie zwischen dem Carbeniumion und dem Iminiumion stabilisiert. Das Ion fungiert in der Reaktion als aminomethylierendes Agens unter elektrophiler Substitution geeigneter nucleophiler Reaktionspartner, wie z.B. der Enol-Form des Ketons. Die so erhaltenen b-Aminoketone bezeichnet man oft auch als Mannichbase. Verwendet man statt dem Salz eines sec. Amins, das eines prim. Amins oder Ammoniak, kompliziert sich der Reaktionsmechanismus dadurch, daß die nun in der Mannichbase enthaltenen Wasserstoffatome an der Reaktion teilnehmen können und zu unerwünschten Produkten führen können. Kondensiert man Amide mit Formaldehyd, erhält man Hydroxymethylamide, die in saurer Umgebung zu Acylmethyleniminiumsalze, mit denen man eine a-Amidomethylierung durchführen kann. Verwendet man statt Formaldehyd ein anderes Aldehyd zur Herstellung des Iminiumsalzes, spricht man von einer a-Amidoalkylierung, durch die man ein weiteres Mittel zur Erzeugung einer C-C-Einfachbindung zur Verfügung hat.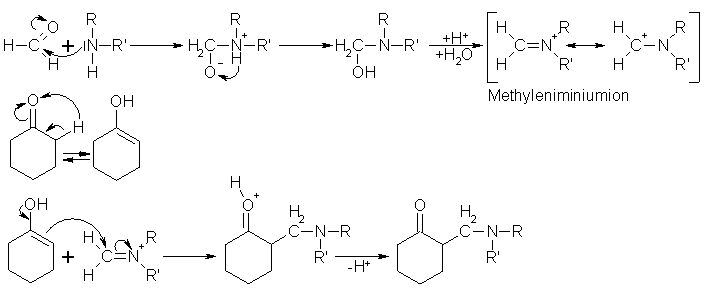 Beyer/Walter
S.544
Beyer/Walter
S.544
Organikum S.484
Vollhardt S.952
Vorlesung
Die McLafferty-Umlagerung kann oft im Massenspektrometer beobachtet werden. Dazu muß eine organische Verbindung in
g-Stellung zu einer CH-Gruppe eine Doppelbindung, wie z.B. >C=O oder >C=NR, tragen. Die Umlagerung verläuft dann über einen 6gliedrigen, cyclischen Übergangszustand, bei dem ein Proton auf das ungesättigte Ladungszentrum übertragen wird. Anschließend eliminiert sich ein Alken.Beyer/Walter S.34
Organikum S.112
Vollhardt S.909
Bei der McMurry-Reaktion werden Alken aus Aldehyden oder Ketonen gebildet, ohne Isolierung des entstehenden Diols. Als Reaktionsauslöser wird fein verteiltes Titan verwendet, welches aus Titantrichlorid und Lithiumaluminiumhydrid oder Kalium zugänglich ist. Die reduktive Kupplung von 2 Molekülen des Ketons oder Aldehyds verläuft hierbei über ein Radikalanion. Die darauf folgende Bildung des Alkens führt man auf die hohe Sauerstoffaffinität des Titans zurück.
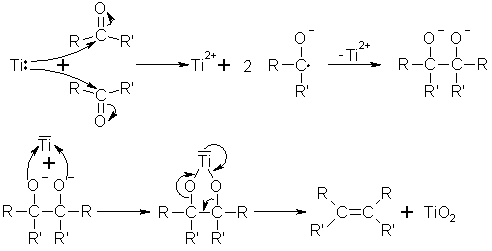
Beyer/Walter S.218
Vollhardt S.732
Meerwein-Ponndorf-Verley-Reduktion
Mit dieser Reduktion läßt sich ein Aldehyd reduktiv in einen prim. Alkohol überführen. Dabei dient als Reduktionsmittel Aluminiumisopropylat in Isopropylalkohol, welches dann mit einem Aldehyd zur Reaktion gebracht wird. Dabei entsteht in einem Redoxprozeß der, dem Aldehyd entsprechende prim. Alkohol und Aceton. Diese Reduktion ist chemoselektiv, das heißt, daß nur >C=O-Gruppen in Aldehyden oder Ketonen durch diese Reaktion reduziert werden, andere reduzierbare Gruppen, wie z.B. C=C-Doppelbindungen, bleiben unangetastet. Die Umkehrreaktion wäre die Oppenauer-Oxidation.
Beyer/Walter S.196
Organikum S.504
Vorlesung
Die Meerwein-Schuster-Reaktion stellt eine Arylierung von
a,b-ungesättigten Carbonylverbindungen oder Carbonsäuren, und deren Derivaten, dar. Die Reaktion verläuft über einen, durch Kupfer(I)- und Kupfer(II)-salze katalysierten, radikalischen Mechanismus. Zur Arylierung verwendet man aromatische Diazoniumsalze. Dabei kann es sowohl zu einer Addition des aromatischen Restes und des Halogenatoms an die Doppelbindung, als auch zu einer Substitution eines Wasserstoffatoms der Doppelbindung durch den Arylrest kommen. Verwendet man ein ungesättigte Carbonsäure decarboxyliert das Produkt zu einem Stilbenabkömmling.Beyer/Walter S.586
Die Michael-Addition ist eine nucleophile Addition von Verbindungen mit aktivierter Methylengruppe (Michael Donoren), z.B. Malonsäure oder Acetessigester, an aktivierte C=C-Doppelbindungen (Michael Akzeptoren), z.B.
a,b-ungesättigte Carbonylverbindungen, Carbonsäureester oder Nitrile, in Gegenwart von basischen Katalysatoren, z.B. Piperidin, Diethylamin oder Natriumalkoholat. Das Reaktionsprodukt kann man leicht hydrolisieren. Dabei entsteht unter Abspaltung von Alkohol und Decarboxylierung eine d-Ketosäure.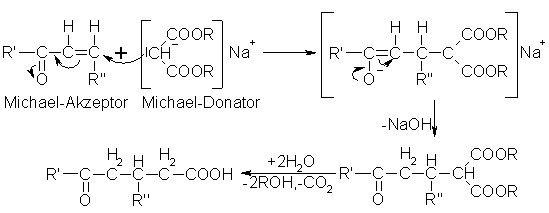
Beyer/Walter S.329
Organikum S.527
Vollhardt S.789
Vorlesung
Diese Reaktion ist eine gute Möglichkeit zur Knüpfung von Kohlenstoff-Phosphor-Bindungen. Dabei läßt man ein Alkylphosphit mit einem Alkylhalogenid reagieren. Dabei wird ein Alkanphosphonsäureester gebildet.
Beyer/Walter S.175
Organikum S.215
Bei der Müller-Rochow-Synthese wird Dichlordimethylsilan direkt aus Methylchlorid in der Dampfphase bei 350°C hergestellt. Dazu wird das Methylchlorid über fein gepulvertes Silicium in Gegenwart von Kupfer als Katalysator geleitet. Dabei entsteht Dichlordimethylsilan.
Beyer/Walter S.180
Die Nef-Reaktion ist eine Aci-Nitroalkan-Spaltung. Bei dieser Reaktion wird ein Nitroalkan in ein Aldehyd oder Keton überführt. Dazu benötigt man ein Salz der Nitroverbindung, welches durch Behandeln der Verbindung mit Natronlauge unter Wasserabspaltung zugänglich ist, und 50% Schwefelsäure. Die Schwefelsäure protoniert die Nitroverbindung, die dann Wasser und Distickstoffoxid (Lachgas) abspaltet, und in das entsprechende Aldehyd oder Keton übergeht.
Beyer/Walter S.157
Vorlesung
Bei der Nesmejanow-Reaktion wird eine Diazoniumsalzlösung mit Quecksilber(II)-chlorid versetzt, wird ein Doppelsalz gefällt. Dieses Doppelsalz wird dann in Aceton suspendiert. Diese Suspension wird dann mit Kupferpulver versetzt und erhitzt. Dabei geht das Doppelsalz in ein Arylquecksilberchlorid über, welches das Endprodukt der Nesmejanow-Reaktion darstellt.
Beyer/Walter S.585
Diese Oxidation kann als Umkehrung der Meerwein-Ponndorf-Verley-Reduktion aufgefaßt werden. Als Edukte werden hierbei sec. Alkohole und ein Keton, z.B. Cyclohexanon, im Überschuß verwendet. In Gegenwart von Aluminium-tert.-butylat entsteht in der Hitze durch einen Redoxprozeß das dem Alkohol entsprechende Keton und der dem Keton entsprechende Alkohol.
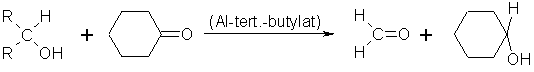
Beyer/Walter S.211
Organikum S.504
Durch die Paal-Knorr-Synthese sind sowohl Pyrrol-Derivat, als auch Furan-Derivate zugänglich. Für die Pyrrolsynthese wird ein 1,4-Diketon mit Ammoniak oder einem prim. Amin umgesetzt. Unter Wasserabspaltung kommt es zu einem Ringschluß, der zu 2,5-Dialkyl- bzw. 2,5-Diarylpyrrole führt. Bei prim. Aminen als 2. Edukt, gibt es N-substituierte Pyrrol-Derivate. Für die Synthese der Furanabkömmlinge werden 1,4-Ketoverbindungen mit wasserentziehenden Mitteln, wie Zinkchlorid oder Phosphor(V)-oxid, erhitzt. Dabei kommt es zum Schluß des Furanringes, der sich leicht hydrolytisch wieder zum Keton, in essigsaurer Lösung durch Zugabe von wenig Schwefelsäure, öffnen läßt.
 Beyer/Walter
S.706 und S.719
Beyer/Walter
S.706 und S.719
Vollhardt S.1144
Vorlesung
Führt man die Photolyse eines Ketons in einem Alken als Lösungsmittel durch, kann das photolytisch angeregte Keton mit dem Alken reagieren. Als Produkt entsteht ein Oxetanderivat. Diese Synthese heißt Paterno-Büchi-Reaktion. Führt man die Reaktion mit Aceton in Cyclopenten durch, folgt eine (2+2)-Cycloaddition zu dem entsprechenden Oxetanderivat.
Beyer/Walter S.225
Die Perkin-Synthese dient zur Synthese von ungesättigten araliphatischen Monocarbonsäuren (aromatische Monocarbonsäure mit ungesättigter Seitenkette), z.B. Zimtsäure. Für die Synthese geht man von aromatischen Aldehyden (Carbonylkomponente) und Acetanhydrid als Edukte aus. In Gegenwart von basischen Kondensationsmitteln, z.B. Natriumacetat oder Pyridin, kommt es zu einem aldolartigen Zwischenprodukt. Das Zwischenprodukt geht unter Wasserabspaltung und anschließender Hydrolyse in die araliphatische Carbonsäure und Essigsäure über. Als erster Reaktionsschritt bildet sich aus dem Acetanhydrid und Natrium-acetat im Gleichgewicht ein Säureanhydridanion (Methylenkomponente), welches nun nucleophil die Carbonylkomponente (im Fall von Zimtsäure ist es Benzaldehyd) angreift. Das nun entstandene Anion wird durch die intermediär gebildete Essigsäure protoniert. Durch das noch vorhandene Acetanhydrid spaltet sich Wasser aus dem Zwischenprodukt ab, dabei wird ein konjugiertes Bindungssystem gebildet. Durch anschließende Hydrolyse erhält man die araliphatische Monocarbonsäure und Essigsäure.
Beyer/Walter S.556
Organikum S.472
Vorlesung
Hierbei wird aus einer Carbonylverbindung mit a-ständiger, acider CH-Gruppe ein Alken (Olefin) synthetisiert. Das ganze geschieht durch Aktivierung der a-ständigen, aciden CH-Gruppe durch eine Trimethylsilylgruppe, die aus Trimethylsilanen und tert.-Butyl-Lithium zugänglich ist. Die Peterson-Olefinierung ist eine Alternative zur Wittig-Reaktion und zur Horner-Emmons-Reaktion. Sie wird dann angewandt, wenn die Alkene durch andere Reaktionen nur schwer zugänglich sind. Der Mechanismus der Peterson-Olefinierung verläuft auf analogem Weg, wie die beiden oben genannten Reaktionen.
Beyer/Walter S.181
Bringt man Alkene und organische Persäuren in indifferenten Lösungsmitteln zur Reaktion, bildet sich das entsprechende Oxiran (Epoxid) und die, der Persäure entsprechende, Carbonsäure. Die Prileschajew-Reaktion verläuft stereospezifisch. So wird aus (E)-Cycloocten nur das cis-Epoxid und aus (Z)-Cycloocten nur das trans-Epoxid gebildet.
Beyer/Walter S.73
Organikum S.271
Vorlesung
Die Prins-Reaktion wird zur Synthese von 1,3-Diolen verwendet. Dabei handelt es sich um eine säurekatalysierte Anlagerung von Formaldehyd an Alkene im wäßrigen Milieu.
Beyer/Walter S.104
Organikum S.468
Die Pschorr-Synthese liefert aus den Edukten o-Nitrobenzaldehyd und Phenylessigsäure als Produkt Phenanthren. Dabei kondensieren die Edukte in einer Perkin-Synthese zunächst zu a-Phenyl-o-nitrozimtsäure. Aufgrund der Abstoßung der Phenyl- und der Carboxylgruppe befinden sich die beiden Phenylkerne in cis-Stellung zueinander, was die Möglichkeit zur Verknüpfung der beiden Ringe gibt. Als nächstes wird die Nitrogruppe katalytisch zu einer Aminogruppe reduziert, wobei die a-Phenyl-o-aminozimtsäure gebildet wird. Setzt man diese weiter mit salpetriger Säure und Salzsäure um, kommt es unter Diazotierung der Aminogruppe zur Bildung des Diazoniumsalzes. Das Diazoniumsalz reagiert in Gegenwart von Kupferpulver unter Ringschluß und Stickstoffentwicklung zur Phenanthren-9-carbonsäure. Die Phenanthren-9-carbonsäure spaltet bei der Vakuumdestilation CO2 ab und bildet Phenanthren. Variiert man die Edukte, lassen sich auch Derivate von Phenanthren synthetisieren.
Beyer/Walter S.645
Wirkt ein Säureanhydrid auf Sulfoxide ein, kommt es zu einer Pummerer-Umlagerung, die als Endprodukt zu a-Acyloxysulfiden führt. Es handelt sich hierbei um eine innere Redoxreaktion. Das Säureanhydrid greift im ersten Schritt der Reaktion das Sauerstoffatom im Sulfoxid an. Das so gebildete Zwischenprodukt überträgt auf das entstandene Säureanion ein Proton, dabei entsteht das Sulfoniumylid. Aus dem Sulfoniumylid wird der enthaltene Säurerest gelöst. Daraus bildet sich ein durch Mesomerie stabilisiertes Carbeniumion, an das sich das Säureion zum Endprodukt addiert. Sie ist für die präperative Chemie sehr von Bedeutung, da durch die Pummerer-Umlagerung eine „geschützte Aldehydgruppe" entsteht.
Beyer/Walter S.150
Im Raschig-Verfahren wird Chlorbenzen aus Benzen, Salzsäure und Luft (es wird der Luftsauerstoff benötigt) hergestellt. Es handelt sich hierbei um eine Oxychlorierung. Die Reaktion wird durchgeführt, indem man die 3 Edukte in der Dampfphase über einen Kupferkontakt leitet.
Beyer/Walter S.481
Mit dieser Methode lassen sich b-Hydroxycarbonsäureester herstellen, die unter Wasseraustritt in den entsprechenden a,b-ungesättigten Carbonsäureester übergehen. Für diese Reaktion läßt man eine Organozinkverbindung eines a-Halogensäureesters in einem indifferenten Lösungsmittel an die C=O-Doppelbindung von Aldehyden oder Ketonen addieren. Die Organozinkverbindung erhält man analog zu Grignard-Verbindungen, wenn man den a-Halogensäureester mit elementarem Zink in Ether oder Benzen zur Reaktion bringt. Dieser Schritt kann mit einer kleinen Menge Iod katalysiert werden. Versetzt man diese Lösung jetzt mit der Carbonylverbindung reagiert das Gemisch ähnlich einer Grignard-Reaktion. Der Zinkhalogenidteil addiert sich an das Carbonylsauerstoffatom, der Säurerest addiert sich an das positivierte C-Atom (aus der Carbonylgruppe). Wird diese Zwischenstufe hydrolysiert, entsteht ein Zinkhydroxyhalogenid und ein b-Hydroxycarbonsäureester, der unter Wasserabspaltung in den a,b-ungesättigten Carbonsäureester übergehet. Man verwendet in dieser Reaktion die Organozinkverbindungen, da diese im Gegensatz zu den Organomagnesiumverbindungen reaktionsträger sind und die Estergruppe nicht angreifen.
Beyer/Walter S.279
Organikum S.521
Vorlesung
Mit der Reimer-Tiemann-Synthese kann man in sehr geringen Ausbeuten Phenolaldehyde synthetisieren. Als Edukte verwendet man Alkalilauge (NaOH; LiOH; usw.), Chloroform und z.B. Phenol. Die 3 Edukte werden bei ca. 65-70°C miteinander zur Reaktion gebracht. Die Lauge überführt das Phenol in ein durch Mesomerie stabilisiertes Phenolation, außerdem reagiert die Lauge mit dem Chloroform über ein Carbanion zum Dichlorcarben (Dichlormethylen). Das Carben lagert sich mit seiner Oktettlücke (es ist eine Elektronenmangelverbindung) an die mesomere Ketoform des Phenolations an. Anschließend kommt es unter Rearomatisierung zu einer Protonenwanderung, dabei bildet sich eine Dichlormethylgruppe aus. Die Dichlormethylgruppe wird im alkalischen Medium zur Aldehydgruppe verseift. Die Reimer-Tiemann-Synthese wird aufgrund der schlechten Ausbeuten nur noch dann angewandt, wenn man einen o-Hydroxyaldehyd synthetisieren möchte, da er dort als Hauptprodukt entsteht.
Beyer/Walter S.539
Organikum S.256
Vorlesung
Die Carbonylierung nach Reppe ist eine Druckreaktion zwischen Acetylen (Ethin) und Kohlenmonoxid. Man benötigt für die Reaktion außerdem noch eine Verbindung mit einem aciden Wasserstoffatom (Alkohole, Wasser) und als Katalysator ein Metallcarbonyl, hauptsächlich Ni(CO)4. Als Produkt erhält man hierbei ungesättigte Carbonsäuren (H2O) oder Carbonsäureester (HO-R).
Beyer/Walter S.101
Vorlesung
Die Cyclisierung ist eine Möglichkeit Benzen herzustellen, sie wird aber aufgrund der Explosionsgefahr von Acetylen unter Druck und erhöhten Temperaturen nicht mehr angewandt. Die Reaktion wird mit Acetylen und selektiv wirkender Katalysatoren durchgeführt. Dabei entstehen Cyclopolyalkene der allgemeinen Formel C2nH2n, wobei n³ 3 sein muß. Läßt man Ethin trimerisieren, wird in geringen Mengen Benzen gebildet. Die Nebenprodukte sind Biphenyl, Naphthalin und andere aromatische Kohlenwasserstoffe.
Beyer/Walter S.100
Vorlesung
Bei dieser Reaktion dient Kupferacetylid (Kupfercarbid; Cu2C2) als Kontaktmittel. Als Edukte können Ethin oder seine monosubstituierten Derivate und Carbonylverbindungen (Aldehyde, Ketone) verwendet werden. Dabei wird das Ethin oder ein monosubstituiertes Derivat, unter Erhaltung der Cº C-Dreifachbindung, an das C-Atom der Carbonylverbindung addiert. Bei Ethin selbst kann es zu 2 Additionen der Carbonylverbindung kommen. Als Produkt erhält man bei den monosubstituierten Derivaten ein Alkinol, bei Acetylen ein Alkinol und/oder ein Alkindiol. Bei den höheren Aldehyden oder Ketonen wird vornehmlich das Alkinol gebildet.
Beyer/Walter S.99
Vorlesung
Bei der Reppe-Vinylierung benötigt man, genau wie bei der Reppe-Ethinylierung, Ethin oder seine monosubstituierten Derivate als Ausgangsstoff. Es erfolgt diesmal eine Addition an eine organische Verbindung, die eine funktionelle Gruppe mit einem beweglichen Wasserstoffatom trägt (=NH; -OH; -SH; -NH2; -CONH2; -COOH). Im Gegensatz zur Reppe-Ethinylierung geht die Cº C-Dreifachbindung in eine Vinylgruppe (C=C-Doppelbindung) über. Der Reaktionsmechanismus wird nun am Beispiel eines Alkohols dargestellt. Die Reaktion wird durch das, dem zu addierenden Alkohol entsprechende, Alkoholation katalysiert, wobei das Alkoholation nucleophil an die Cº C-Dreifachbindung addiert. Das so entstandene Carbanion reagiert mit einem Molekül Alkohol, unter Freisetzung des Alkoholations, zum Vinylether. Je nach Edukt kann es zu verschiedenen Endprodukten kommen, z.B. Alkohole ergeben Vinylether; Carbonsäuren ergeben Vinylester.
Beyer/Walter S.99
Vorlesung
Dieses Verfahren wird zur technischen Herstellung von Acetylen verwendet. Dabei wird Methan mit Sauerstoff unvollständig bei 1500°C verbrannt. Schreckt man dann das heiße Gasgemisch mit Wasser ab, erhält man in 8-9% Ausbeute Ethin, der Rest ist Synthesegas (H2; CO). Trotz der geringen Ethinausbeute ist die Energieausbeute mit 75% sehr gut.
Beyer/Walter S.93
Mit der Sandmeyer-Reaktion wird ein weiterer Weg zur Herstellung von Chlor- und Brombenzen dargestellt. Um die Sandmeyer-Reaktion durchzuführen läßt man eine wäßrige, aromatische Diazoniumsalzlösung in eine warme Kupfer(I)-chlorid, bzw. -bromid, Lösung, die mit Salzsäure, bzw. Bromwasserstoffsäure, angesäuert wurde, fließen. Das Kupfer(I)-halogenid dient als Katalysator. Es bildet mit dem Diazoniumion einen „Primärkomplex", der in der Wärme Stickstoff abspaltet. Dabei kommt es zu einer Einelektronenübertragung vom Kupfer(I) zum Stickstoff, wobei das Kupfer(I) in Kupfer(II) übergeht. Das Kupfer(II)-halogenid wird durch das intermediär gebildete Phenylradikal wieder zum Kupfer(I)-halogenid reduziert und es entsteht das Halogenbenzen. Ein Sonderfall der Sandmeyer-Reaktion ist die Substitution der Diazogruppe durch die Cyangruppe. Als Edukte werden hier eine Diazoniumsalzlösung und mit Kaliumcyanid komplex gelöstes Kupfer(I)-cyanid (K3[Cu(CN)4]). Als Produkt entsteht hierbei Benzonitril.
Beyer/Walter S.585
Organikum S.566
Vollhardt S.1019
Durch die Schiemann-Reaktion sind Fluorarene zugänglich. Die Reaktion verläuft ähnlich der Sandmeyer-Reaktion. Die Fluorierung ist durch die thermische Zersetzung von Diazoniumtetrafluorboraten möglich. Hierbei kann auf eine Zugabe eines Katalysators verzichtet werden. Um die Fluorierung zu erreichen gibt man zu der Lösung eines aromatischen Diazoniumsalzes HBF4. Hierdurch wird das Diazoniumtetrafluorborat gebildet. Wenn man dieses Salz auf ca. 120°C erwärmt,wird Stickstoff und BF3 abgespalten und es entsteht das entsprechende Fluoraren.
Vollhardt S.1020
Mit der Schmidt-Reaktion werden prim. Amine aus Carbonsäuren, Stickstoffwasserstoffsäure (N3H) und konzentrierter Schwefelsäure als Katalysator. Die Stickstoffwasserstoffsäure addiert sich nucleophil an das C-Atom der Carboxygruppe. Es spaltet sich Stickstoff und durch die Schwefelsäure Wasser ab. Es kommt nun zu einer nucleophilen Umlagerung zum Isocyanat, welches mit Wasser unter Kohlendioxidentwichlung (Kettenverkürzung) in das entsprechende prim. Amin übergeht.
Beyer/Walter S.163
Organikum S.595
Vollhardt S.950
Die Reaktion verläuft genau wie das Einhorn-Verfahren. Es wird nur mit wäßrigem Alkali (z.B. NaOH), statt mit Pyridin, als 2. Edukt gearbeitet.
Beyer/Walter S.549
Aus Tosylhydrazonen, die aus Tosylhydrazin (H3C-C6H4-SO2-NHNH2) und einer Carbonylverbindung zugänglich sind, und einem Überschuß an sehr starker Base, z.B. n-Butyllithium, wird ein Alken hergestellt. Die Reaktion verläuft über ein Dianion.
Beyer/Walter S.213
Hierbei wird ein Alken, Diiodmethan und überschüssiges aktiviertes Zink in wasserfreiem Diethylether umgesetzt. Dabei entsteht, aus dem Diiodmethan und dem Zink Iodmethyl-Zinkiodid, welches mit dem Alken, in einem bicyclischen Übergangszustand, zu einem Cyclopropanderivat weiter reagiert. Die Simmons-Smith-Reaktion verläuft stereospezifisch, d.h. aus cis-Alkenen entstehen cis-Cyclopropane und aus trans-Alkenen werden trans-Cyclopropane.
Beyer/Walter S.189
Vollhardt S.961
Die Skraupsche-Synthese ist ein Verfahren zur Produktion von Chinolin. Dazu erhitzt man Anilin mit wasserfreiem Glycerol, konzentrierter Schwefelsäure, Eisen(II)-sulfat und Nitrobenzen, als Dehydrierungsmittel. Im ersten Schritt wird Glycerol durch Wasserentzug in Acrolein überführt, das mit dem Anilin zum 1,2-Dihydro-chinolin kondensiert. Das Nitrobenzen dehydriert im letzten Schritt das 1,2-Dihydro-chinolin zum Chinolin.
Beyer/Walter S.773
Organikum S.524
Vorlesung
Mit dieser Reaktion wird Benzaldehyd aus Benzylchlorid und Hexamethylentetramin gebildet. Als Reaktionsmedium dient wäßriges Ethanol oder verdünnte Essigsäure bei einem pH zwischen 3,0 und 6,5. Das Benzylchlorid bildet im ersten Reaktionsschritt mit dem Hexamethylentetramin ein quart. Salz, das durch Hydrolyse in Benzylamin, Ammoniak, Ammoniumchlorid und Formaldehyd gespalten wird. Aus dem Ammoniak und dem Formaldehyd bildet sich im folgenden Methylenimin. Das Methylenimin wird beim Ansäuern zu einem Kation protoniert. Durch einen Hydridtransfer (H-) von der CH2-Gruppe des Benzylamins zum Kation des Methylenimins, wird Methylenamin und ein Benzyliminiumkation gebildet. Durch Hydrolyse des Benzyliminiumkations wird das Benzaldehyd und Ammoniak gebildet.
Beyer/Walter S.531
Durch die Stephen-Reduktion können aromatische Nitrile zu den entsprechenden Aldehyden reduziert werden. Dazu leitet man in eine etherische Lösung des Nitrils Chlorwasserstoff ein, und gibt etwas wasserfreies Zinn(II)-chlorid dazu. Aus dem Chlorwasserstoff und dem Zinn(II)-chlorid bildet sich die Tetrachlorzinn(II)-säure (H2[SnCl4]). Die Tetrachlorzinn(II)-säure reduziert das Nitril zu einem Aldiminhexachlorstannat, welches leicht zum Aldehyd hydrolysiert wird. Statt einer etherischen Lösung kann als Lösungsmittel auch Ethylformiat (Ameisensäureethylester) oder Essigsäureethylester verwendet werden.
Beyer/Walter S.532
Die Stobbe-Kondensation wird hauptsächlich dazu verwendet, um in einer aromatischen Carbonylverbindung die Carbonylgruppe durch einen Propionsäurerest (Propansäurerest) auszutauschen. Um dieses Ziel zu erreichen, werden Aldehyde oder Ketone in Gegenwart einer starken Base, z.B. Kalium-tert.-butylat, mit einem Bernsteinsäurediester umgesetzt. Die Reaktion führt über ein Aldol zum Kaliumsalz des Alkylidenbernsteinsäurehalbester, der unter saurer Hydrolyse, am Besten eignet sich Bromwassestoffsäure in Eisessig, unter Decarboxylierung in ein g-Lacton bzw. in eine b,g-ungesättigte Monocarbonsäure übergeht. Beide Produkte lassen sich katalytisch zu der gleichen gesättigten Monocarbonsäure reduzieren.
Beyer/Walter S.332
Die Strecker-Synthese ist ein Syntheseweg für a-Aminosäuren. Dazu wird im ersten Reaktionsschritt Blausäure an ein Aldehyd addiert, wobei ein Cyanhydrin entsteht, das mit Ammoniak unter Wasserabspaltung zu einem Aminonitril weiter reagiert. Hydrolysiert man das Aminonitril mit konzentrierter Mineralsäure, entsteht die dem Aldehyd entsprechende a-Aminosäure und Ammoniak.
Beyer/Walter S.286
Organikum S.463
Vollhardt S.1090
Vorlesung
Die Swarts-Reaktion ist eine schonende Möglichkeit für die Synthese von fluorierten Alkanen. Da eine direkte Fluorierung von Alkanen sehr exotherm ist, und meist explosionsartig verläuft. Als Fluorierungsmittel dienen hierbei die weniger reaktionsfähigen Metallfluoride, wie z.B. Silber-, Quecksilber(I)- oder Kaliumfluorid. Die Metallfluoride reagieren mit einem Alkylhalogenid in einer Substitutionsreaktion zum Alkylfluorid und dem Metallhalogenid.
Beyer/Walter S.139
Vorlesung
Durch Dimethylsulfoxid (DMSO), das mit Oxalylchlorid aktiviert wird, in Methylenchlorid (CH2Cl2) werden prim. Alkohole zum entsprechenden Aldehyd oxidiert. Als Oxidationsmittel dient das, aus den primären Edukten DMSO und Oxalylchlorid unter spontaner Decarboxylierung und Decarbonylierung (-CO) gebildete, sauerstofffreie, aktivierte Zwischenprodukt. Dieses Zwischenprodukt reagiert mit dem prim. Alkohol zu einem Alkoxysulfoniumsalz, das in Gegenwart einer Base, z.B. Triethylamin, ein Proton abgibt und somit zum Aldehyd und Dimethylsulfid zerfällt.
Beyer/Walter S.194
Vorlesung
Diese Reaktion ist eine Abwandlung der Cannizzaro-Reaktion. Die Tischtschenko-Reaktion disproportioniert aber im Gegensatz dazu auch aliphatische Aldehyde. Als Kondensationsmittel werden hier Aluminiumalkoholate verwendet, die eine Reaktion nur in einem wasserfreien Medium erlauben, da sie sich
mit Wasser zu Aluminiumhydroxid und dem Alkohol zersetzen. Als Lösungsmittel wird darum der dem Alkoholat entsprechende absolute Alkohol verwendet. Die entstehende Carbonsäure verestert mit dem Alkohol sofort. Darum entsteht als Endprodukt der Carbonsäureester.
Beyer/Walter S.202
Die Traube-Synthese ist für die Herstellung von Harnsäure und deren Derivate von Bedeutung. Sie wird im folgenden durch die Synthese von Harnsäure dargestellt. Man kondensiert, in diesem Fall wird Ethanol abgespalten, zu diesem Zweck Harnstoff mit Cyanessigester zum Cyanacetylharnstoff, der durch eine Base, z.B. Natronlauge, zum 6-Aminouracil cyclisiert wird. Durch salpetrige Säure wird das 6-Aminouracil unter Wasserabspaltung in das 6-Amino-5-nitrosouracil überführt. Durch Reduktion der Nitrogruppe entsteht das 5,6-Diaminouracil, welches mit Chlorameisensäureethylester zu einem Urethan reagiert. Das Urethan spaltet in der Hitze Ethanol ab und geht in die Harnsäure über.
Beyer/Walter S.798
Diese Reaktion stellt eine nucleophile Substitution an einem aromatischen Heterocyclus, z.B. Pyridin, dar. Dabei bringt man das Pyridin mit Natriumamid (NaNH2) in flüssigem Ammoniak zur Reaktion. Dabei greift die NH2-Gruppe nucleophil in o-Stellung zum Heteroatom den aromatischen Kern an. Die o-Stellung wird dabei bevorzugt, da sie am Besten durch Mesomerie stabilisiert ist. Der Unterschied zu einer elektrophilen Substitution ist, daß es zu keiner Deprotonierung kommt, sondern ein Hydridion abgespalten wird. Das Hydridion greift nun an der NH2-Gruppe an, wobei ein 2-Pyridinamidanion und ele
mentarer Wasserstoff entsteht. Ähnliche Reaktionen treten mit Organolithium- und Grignard-Verbindungen auf.
Beyer/Walter S.763
Vollhardt S.1157
Das Twitchell-Verfahren ist eine Möglichkeit zur sauren „Verseifung" von Fetten. Die im Gegensatz zur alkalischen „Verseifung" einen Emulgator, das Twitchell-Reaktiv, benötigt, da hier keine Seifen (Natriumsalze der Fettsäuren) entstehen. Das Twitchell-Reaktiv wird aus Ölsäure und Naphthalin mit konzentrierter Schwefelsäure hergestellt. Diese Reagenz wird in ½-1% der Fettmenge zugegeben. Läßt man dieses Gemisch 24 Stunden bei 100°C reagieren, kommt es zu einer 90% Spaltung des Fettes in die freie Fettsäure und Glycerol.
Beyer/Walter S.248
Triphenylamin [(C6H5)3N] wird durch Erhitzen von Diphenylamin und Iodbenzen mit Kaliumcarbonat und etwas Kupferbronze in Nitrobenzen dargestellt. Die Ullmann-Reaktion führt als Nebenprodukt noch zu Kaliumiodid und Kaliumhydrogencarbonat.
Beyer/Walter S.580
Durch diese Reaktion wurde eine Formylierung von aromatischen Verbindungen in guten Ausbeuten realisiert. Als besonders gute Edukte haben sich Phenolether und Dialkylaniline erwiesen. Der Mechanismus dieser Reaktion kann auch als Spezialfall der Friedel-Crafts-Acylierung aufgefaßt werden. Als Edukte verwendet man außer dem Phenolether noch N-Methylformanilid, welches durch N-Methyl- oder N,N-Dimethylformamid ersetzt werden kann, und Phosphorchloridoxid. Das N-Methylformanilid reagiert mit dem Phosphorchloridoxid ein Chlormethyleniminiumsalz, das als „Vilsmeier-Haack-Komplex" bezeichnet wird. Dieser Komplex greift den aromatischen Kern elektrophil, bevorzugt in p-Stellung zur Ethergruppe, an. Das dabei entstehende Produkt geht bei Hydrolyse in das p-Aldehyd, das N-Methylanilin und HPO2Cl2 über.
Beyer/Walter S.538
Organikum S.343
Vorlesung
Das Wacker-Hoechst-Verfahren ermöglicht die Herstellung von Acetaldehyd aus Ethen, aus den homologen Alkenen werden Ketone gebildet. Die Oxidation findet im wäßrigen Milieu statt und wird mit Palladiumchlorid katalysiert, das mit dem Alken einen p1-Komplex bildet. An diesen Komplex greift ein Hydroxidion nucleophil an, was zur Bildung eines s-Komplexes führt. Durch eine b-Eliminierung wird aus dem s-Komplexe ein p2-Komplex gebildet, der mit Sauerstoff in die Carbonylverbindung übergeht. Das so entstandene Palladium wird durch den Zusatz von Kupfer(II)-chlorid in den Katalysator zurück verwandelt.
Beyer/Walter S.205 und S.384
Vollhardt S.485
Vorlesung
Beim Wacker-Verfahren wird Keten (H2C=C=O) mit Eisessig umgesetzt. Diese Reaktion dient zur technischen Herstellung von Essigsäureanhydrid, da die Edukte zu 95 % in das Produkt übergehen.
Bayer/Walter S.256
Organikum S.299
Vollhardt S.819
Die Wagner-Meerwein-Umlagerung ist eine, durch Säuren katalysierte, nucleophile 1,2-Umlagerung von sec. Alkoholen. Dabei wird primär der Alkohol durch die Mineralsäure zu einem Oxoniumion protoniert. Das Kation spaltet Wasser zu einem Carbeniumion ab. Das Carbeniumion stabilisiert sich unter Deprotonierung und einer Alkylanionenwanderung zu einem Alken. Diese Umlagerung ist in der Reihe der Terpene von besonderer Bedeutung.
Bayer/Walter S.219
Organikum S.589
Vollhardt S.299
Vorlesung
Erhitzt man einen aliphatisch-aromatischen Keton (R1-CO-R2; wobei R1 ein aliphatischer und R2 ein aromatischer Rest ist) mit Schwefel in Morpholin, entsteht ein Carbonsäurethioamid (in diesem Fall ein Thiomorpholid), mit gleicher Kohlenstoffzahl. Die Carbonylgruppe wurde zu einer CH2-Gruppe reduziert. Durch alkalische Hydrolyse wird das Thiomorpholid in eine arylsubstituierte aliphatische Carbonsäure überführt. Der genaue Mechanismus ist noch nicht geklärt, es wird aber vermutet, daß die Reaktion über eine Folge von Reduktionen und Oxidationen entlang der Kohlenstoffkette verläuft.
Beyer/Walter S.546
Organikum S.372
Diese Ethersynthese beruht auf der Reaktion von Alkalialkoholaten mit Alkylhalogeniden. Als Reaktionsmedium dient entweder der, dem Alkoxid entsprechende, Alkohol, Dimethylsulfoxid oder ähnliche polare Lösungsmittel. Ob diese SN2-Reaktion abläuft oder eine E2-Reaktion stattfindet hängt hauptsächlich vom Alkylrest ab. So reagiert. z.B. Iodmethan mit dem Isopropylalkoholation zum SN2-Produkt dem Isopropylmethylether, verwendet man Isopropyliodid und das Methoxylation (Methanolat) wird überwiegend das E2-Produkt Propen gebildet.
Beyer/Walter S.144
Organikum S.211
Vollhardt S.311
Vorlesung
Die Wittig-Reaktion ist eine Carbonylolefinierung, d.h. aus Ketonen wird ein Alken gebildet. Die Olefinierung findet mit Alkylidenphenylphosphoranen [(C6H5)3P=CHR] und einem Keton im alkalischen Medium bei Raumtemperatur statt, wobei Alkene gebildet werden. Zuerst reagieren das Phosphoran und das Keton zu einem Oxaphosphetan, in dem es zu einer Pseudorotation mit anschließender Ringöffnung zum Betain I kommt. Dieses kann unter Triphenylphosphinoxidabspaltung [(C6H5)3O] zu einem (Z)-Alken reagieren, oder durch eine Umlagerung zum thermodynamisch stabileren Betain II reagieren, welches unter (C6H5)3O-Abspaltung zu einem (E)-Alken. Das (Z)-Alken wird bevorzugt gebildet, wenn man von den instabileren (R=Alkyl oder O-Alkyl) Alkylidenphenylphosphoranen ausgeht, da das Betain I dadurch sehr kurzlebig ist und sich nicht weiter zum Betain II umlagern kann. Verwendet man statt dessen Alkylidenphenylphosphorane, die stabilisierende Gruppen, wie Halogene, Arylreste, CHO- oder CN-Gruppen, als Rest R tragen, kommt man zu einem stabileren Betain I, daß sich dann zum Betain II umlagert. Dabei werden dann (E)-Alkene gebildet. Diese Stereochemie ist aber auch noch von den Reaktionsbedingungen, wie Lösungsmittel und Salzgehalt, abhängig.
Beyer/Walter S.177
Organikum S.481
Vollhardt S.734
Vorlesung
Diese Synthese wird als Grundstein der heutigen organischen Chemie verstanden, da es mit ihr zum ersten mal gelang eine pflanzliche, bzw. tierische Substanz aus einem anorganischen Edukt herzustellen. Wöhler dampfte dazu eine wäßrige Ammoniumcyanat-Lösung ein. Dabei entsteht im Gleichgewicht Harnstoff.
Beyer/Walter S.355
Vollhardt S.3
Vorlesung
Mit dem Verfahren nach Wohl ist es möglich Aldosen (Aldehyd-Zucker) in eine Aldose mit einer um ein C-Atom verkürzten Kohlenstoffkette. Um dies zu erreichen wird die Ausgangsaldose mit Hydroxylamin umgesetzt, was den Zucker in ein Aldoxim überführt. Erhitzt man das Aldoxim mit Acetanhydrid entsteht unter Dehydratisierung der Oximgruppe eine vollständig acetyliertes Aldononitril, das in der Hitze mit ammoniakalischer Silberoxidlösung Blausäure abspaltet. Dieses Spaltprodukt geht unter Hydrolyse der Acetylgruppen in die um ein C-Atom ärmere Aldose über.
Beyer/Walter S.437
Vollhardt S.1105
Diese Halogenierung von Alkenen beruht auf einem radikalischen Mechanismus, bei dem man mit kleinen Mengen Halogen arbeiten muß. Als besonders guter Halogenlieferant hat sich hierbei das N-Bromsuccinimid (NBS) herausgestellt. Dazu wird das NBS in Tetrachlorkohlenstoff suspendiert und mit we
nig Bromwasserstoff versetzt. Dadurch wird andauernd eine kleine Menge Brom freigesetzt, das durch den Einfluß von Strahlung in Radikale dissoziiert. Ein Bromradikal abstrahiert im ersten Schritt ein Wasserstoffatom in Allylstellung des Alkens. Der so gebildete Bromwasserstoff setzt wieder Brom aus dem NBS frei, das Allylradikal greift nun an einem Brommolekül radikalisch an und bildet ein Bromalken und ein weiteres Bromradikal. Verwendet man unsymmetrische Alkene, werden unsymmetrische Allylradikale gebildet, die zu Produktgemischen führen.
Beyer/Walter S.331
Vollhardt S.556
Vorlesung
Diese Reduktion überführt eine Carbonylgruppe in eine CH2-Gruppe. Die Wolff-Kishner-Reduktion findet im Gegensatz zur Clemmensen-Reduktion in einer alkalischen Umgebung statt. Man setzt dazu das Keton- oder Aldehydhydrazon mit einer starken Base,wie Natriumethanolat bei ca. 200°C um. Bei der Reduktion zum Kohlenwasserstoff kommt es zu Stickstoffentwicklung.
Beyer/Walter S.217
Organikum S.447
Vollhardt S.729
Vorlesung
In Gegenwart von Metallkatalysatoren, wie kolloides Silber oder Kupfer, und höheren Temperaturen spalten Diazoketone Stickstoff ab und es wird eine Elektronenmangelverbindung, ein Carben gebildet. Dieses Carben stabilisiert sich durch eine anionische Wanderung des Restes an das C-Atom mit dem Elektronensextett unter gleichzeitiger Ausbildung einer C=C-Doppelbindung. Es entsteht so ein sehr reaktives Teilchen, ein Keten. Das Keten wird bei der Arndt-Eisert-Synthese weiter umgesetzt. Der Mechanismus der Wolff-Umlagerung wurde schon bei der Arndt-Eisert-Synthese gezeigt.
Beyer/Walter S.255
Organikum S.590
Die Wurtz-Fittig-Synthese ist eine auf aromatische Kohlenwasserstoffe übertragene Wurtz-Synthese. Dazu wird ein Arylhalogenid mit Natrium in etherischer Lösung zu einer alkalimetallorganischen Verbindung umgesetzt, als Nebenprodukt dieses ersten Schrittes entsteht noch das Natriumhalogenid. In einem 2. Reaktionsschritt wird die alkalimetallorganische Verbindung mit einem Alkylhalogenid umgesetzt. Jetzt entsteht das Endprodukt, ein alkylsubstituierter aromatischer Kohlenwasserstoff. Als Nebenprodukte treten Bialkyle und Biaryle auf. Die Ausbeute vom Hauptprodukt hängt davon ab, ob das aromatisch gebundene Halogenatom schneller als das aliphatisch Gebundene gegen das Natrium ausgetauscht wird. Ferner muß die Arylnatriumverbindung schneller mit dem Alkylhalogenid, als mit dem Arylhalogenid reagieren, das ist meistens der Fall, da gewöhnlich die Kohlenstoff-Halogen-Bindung im Alkylhalogenid leichter polarisierbar ist, als die im Arylhalogenid.
Beyer/Walter S.476
Bei der Wurtz-Synthese werden aus Alkylhalogeniden mit Natrium in absolutem Ether als Lösungsmittel Alkane synthetisiert. Dazu setzt man das Alkylhalogenid mit Natrium um. Als Zwischenprodukt erhält man ein Alkylnatrium, welches mit einem weiteren Molekül Alkylhalogenid zum Alkan und dem Natriumhalogenid reagiert. Verwendet man 2 verschiedene Alkylhalogenide entsteht ein Produktgemisch aus allen Möglichen Produkten, d.h. werden die beiden Alkylhalogenide R1-X und R2-X so behandelt, entstehen als Produkte R1-R1, R1-R2 und R2-R2. Am leichtesten läuft die Reaktion mit Iodiden ab, gefolgt von den Bromiden und Chloriden. Mit Alkylfluoriden kommt es zu keiner Reaktion.
Beyer/Walter S.57
Organikum S.516
![]()